
Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer



Abstract
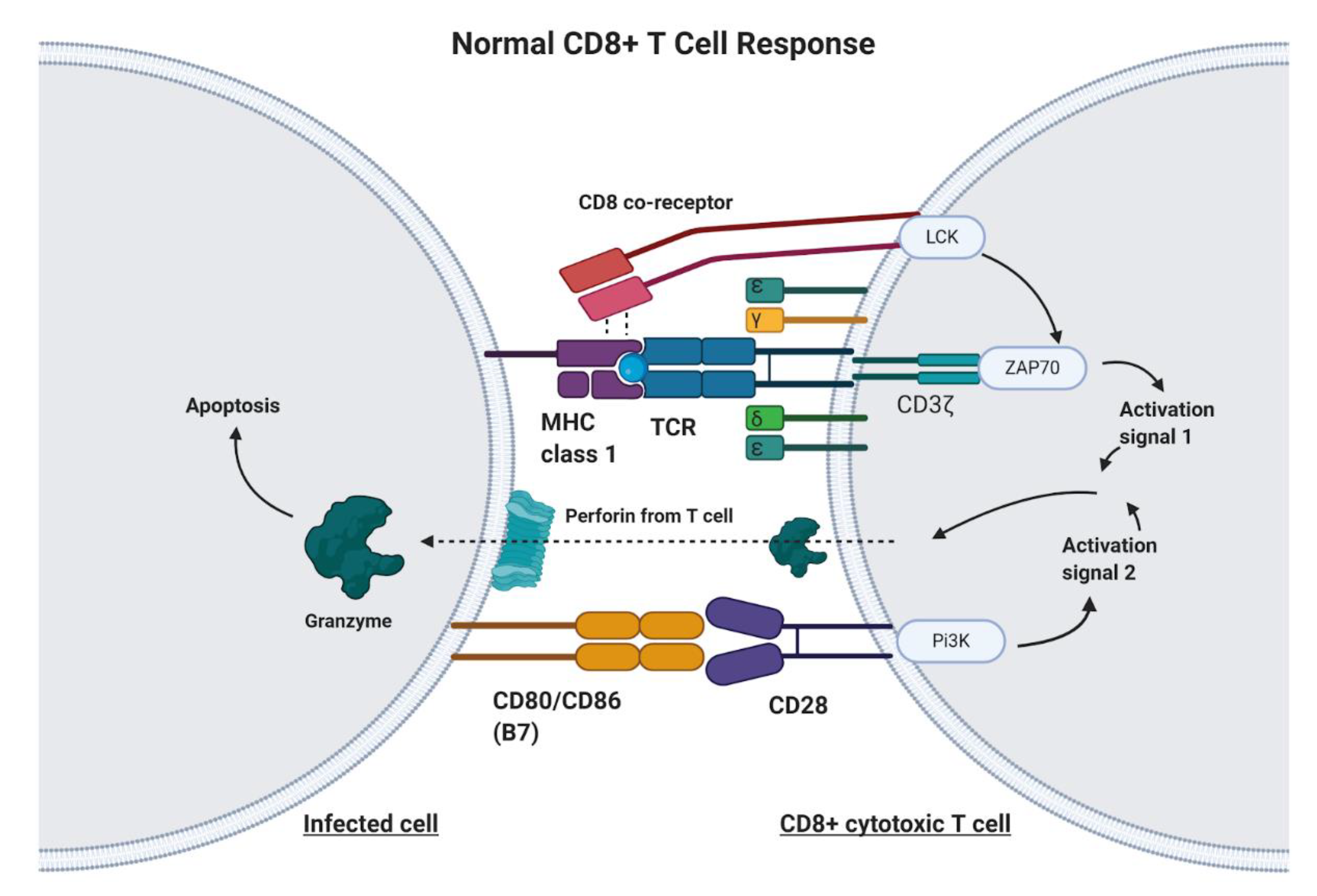
1. Introduction
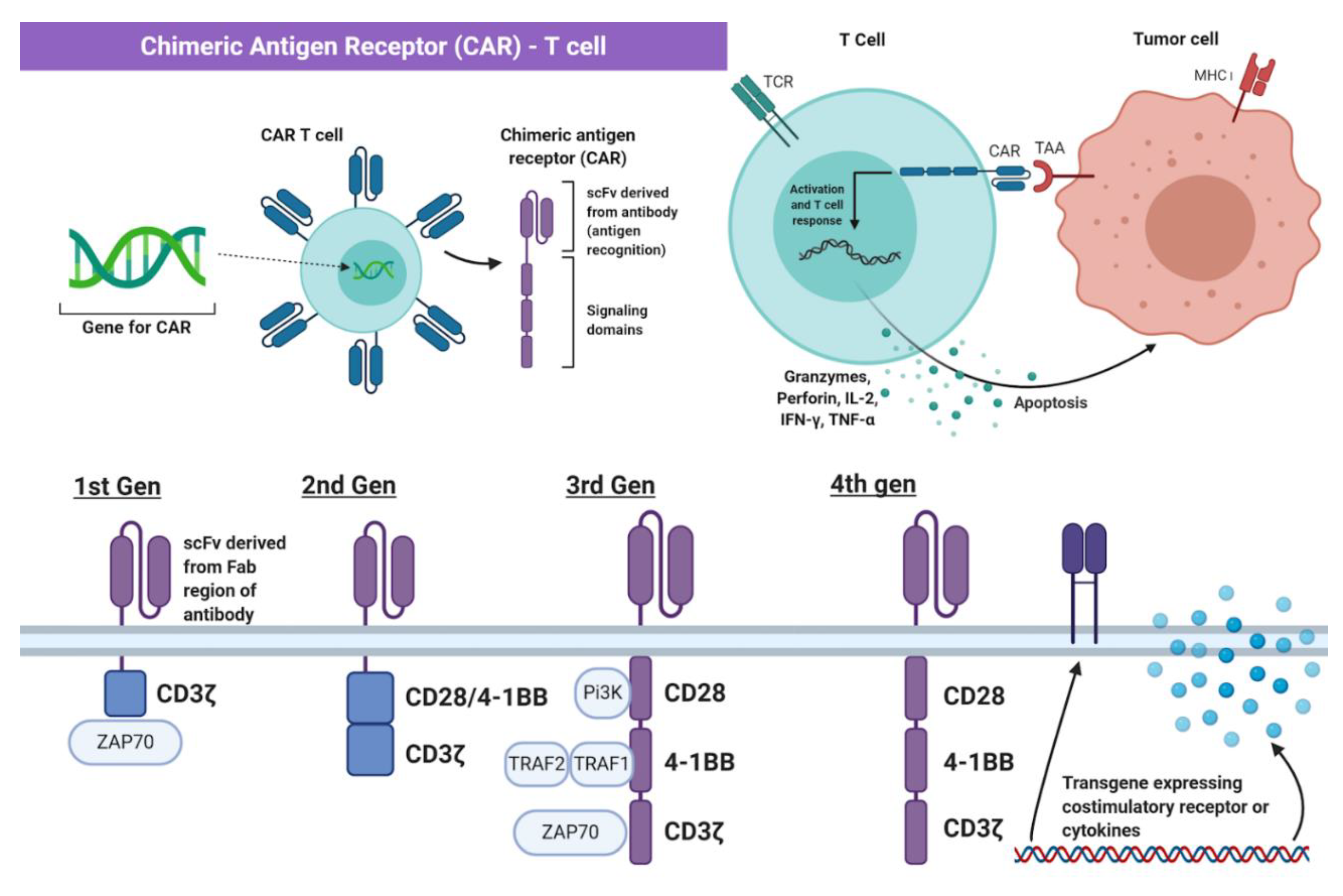
2. Principles of Chimeric Antigen Receptor-T Cell (CAR-T) Therapy
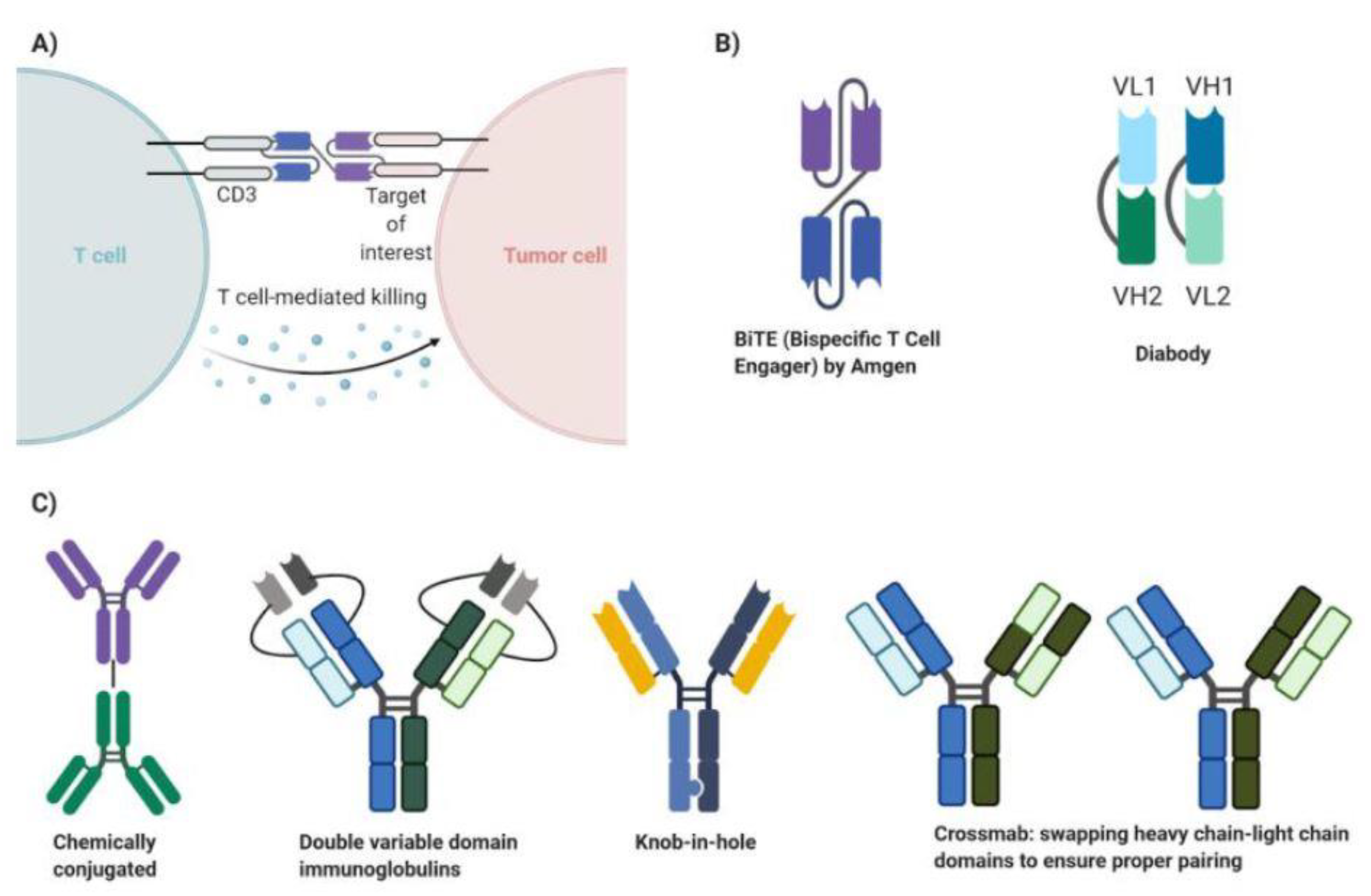
3. Principles of Bispecific Antibody Therapy
4. Development of CAR-T Cell Therapy in Breast Cancer
5. Development of Bispecific Antibody Therapy in Breast Cancer
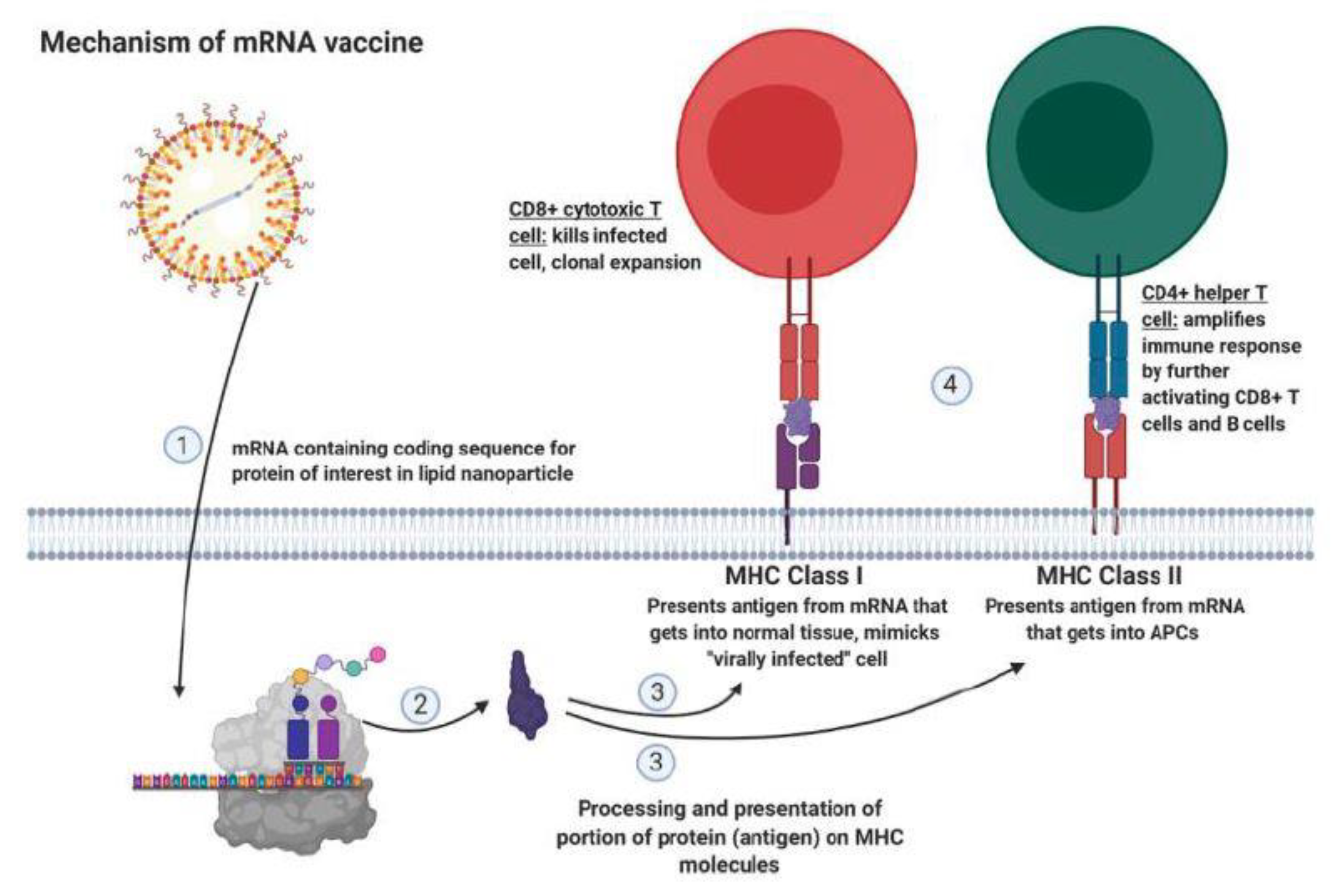
6. Improving Immunotherapy by Modulating Immune Checkpoint, Transforming Growth Factor (TGF), and IL-7R Signaling
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| CAR-T cell | Chimeric antigen receptor-T cell |
| BsAb | Bispecific antibody |
| HER2/ERBB2 | Human epidermal growth factor receptor 2 |
| ER | Estrogen receptor |
| PR | Progesterone receptor |
| EGFR | Epidermal growth factor receptor |
| HR | Hormone receptor |
| TNBC | Triple negative breast cancer |
| MHC | Major histocompatibility complex |
| TCR | T cell receptor |
| scFv | Single-chain variable fragment |
| APC | Antigen presenting cell |
| Fab | Fragment antigen binding |
| PBL | Peripheral blood lymphocytes |
| BiTE | Bispecific T cell engager (by Amgen) |
| Fc | Fragment crystallizable |
| MDA-MB-231 | Human breast cancer cell line |
| HUVEC | Human primary endothelial cell line |
| Hs578T | Human breast cancer cell line |
| MDA-MB-468 | Human breast cancer cell line |
| MCF7 | Human breast cancer cell line |
| MDA-MB-435 | Human breast cancer cell line |
| SKBR-3 | Human breast cancer cell line |
| T47D | Human breast cancer cell line |
| FITC | Fluorescein |
| antigen 1- antigen 2 BsAb (ex: CD3-p95HER2 BsAb) | Format to describe the two molecular targets of the BsAb |
| FITC-Folate BsAb (does not follow conventional BsAb naming format) | BsAb targeting FITC and Folate Receptor (folate targets folate receptor) |
| FITC-DUPA BsAb (does not follow conventional BsAb naming format) | BsAb targeting prostate specific membrane antigen (DUPA targets prostate specific membrane antigen) |
| FITC-AZA BsAb (does not follow conventional BsAb naming format) | BsAb targeting FITC and carbonic anhydrase (AZA targets carbonic anhydrase) |
References
- Breast Cancer Statistics and Resources. Available online: https://www.bcrf.org/breast-cancer-statistics-and-resources (accessed on 22 December 2020).
- American Cancer Society. Cancer Facts and Figures 2020. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html (accessed on 22 December 2020).
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Cato, L.; Jeselsohn, R. Chapter 29—Hormone-Responsive Cancers. In Yen and Jaffe’s Reproductive Endocrinology, 8th ed.; Strauss, J.F., Barbieri, R.L., Eds.; Elsevier: Philadelphia, PA, USA, 2019; pp. 717–741. ISBN 978-0-323-47912-7. [Google Scholar]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Bargmann, C.I.; Hung, M.C.; Weinberg, R.A. The Neu Oncogene Encodes an Epidermal Growth Factor Receptor-Related Protein. Nature 1986, 319, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Hung, M.-C. Overexpression of ErbB2 in Cancer and ErbB2-Targeting Strategies. Oncogene 2000, 19, 6115–6121. [Google Scholar] [CrossRef] [PubMed]
- Joshi, H.; Press, M.F. 22—Molecular Oncology of Breast Cancer. In The Breast, 5th ed.; Bland, K.I., Copeland, E.M., Klimberg, V.S., Gradishar, W.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 282–307. ISBN 978-0-323-35955-9. [Google Scholar]
- Chavez-MacGregor, M.; Mittendorf, E.A.; Clarke, C.A.; Lichtensztajn, D.Y.; Hunt, K.K.; Giordano, S.H. Incorporating Tumor Characteristics to the American Joint Committee on Cancer Breast Cancer Staging System. Oncologist 2017, 22, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3. [Google Scholar]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T Cells: Design Concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Subklewe, M.; Von Bergwelt-Baildon, M.; Humpe, A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus. Med. Hemother. 2019, 46, 15–24. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRzeta /CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric Receptors with 4-1BB Signaling Capacity Provoke Potent Cytotoxicity against Acute Lymphoblastic Leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- CD247 Molecule Homo Sapiens (Human) Gene NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=919#reference-sequences (accessed on 11 January 2021).
- Linsley, P.S.; Ledbetter, J.A. The Role of the CD28 Receptor during T Cell Responses to Antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, M.H.; Turka, L.A. The Role of T-Cell Costimulatory Activation Pathways in Transplant Rejection. N. Engl. J. Med. 1998, 338, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, A.T.C.; Mufti, G.J.; Guinn, B. Role of 4-1BB:4-1BB Ligand in Cancer Immunotherapy. Cancer Gene Ther. 2004, 11, 215–226. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Brocker, T.; Karjalainen, K. Signals through T Cell Receptor-Zeta Chain Alone Are Insufficient to Prime Resting T Lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Curran, K.J. Novel Cellular Therapies for Leukemia: CAR-Modified T Cells Targeted to the CD19 Antigen. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 143–151. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical Manufacturing of CAR T Cells: Foundation of a Promising Therapy. Mol. Ther. Oncol. 2016, 3, 16015. [Google Scholar] [CrossRef]
- Anson, D.S. The Use of Retroviral Vectors for Gene Therapy—What Are the Risks? A Review of Retroviral Pathogenesis and Its Relevance to Retroviral Vector-Mediated Gene Delivery. Genet. Vaccines Ther. 2004, 2, 9. [Google Scholar] [CrossRef]
- Jolly, D.J. Retroviral Vectors. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Academic Press: New York, NY, USA, 2002; pp. 153–166. ISBN 978-0-12-227555-5. [Google Scholar]
- Patel, D.H.; Misra, A. 5—Gene Delivery Using Viral Vectors. In Challenges in Delivery of Therapeutic Genomics and Proteomics; Misra, A., Ed.; Elsevier: London, UK, 2011; pp. 207–270. ISBN 978-0-12-384964-9. [Google Scholar]
- Singh, H.; Huls, H.; Kebriaei, P.; Cooper, L.J.N. A New Approach to Gene Therapy Using Sleeping Beauty to Genetically Modify Clinical-Grade T Cells to Target CD19. Immunol. Rev. 2014, 257, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Wiesinger, M.; Abken, H.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Schaft, N. A GMP-Compliant Protocol to Expand and Transfect Cancer Patient T Cells with MRNA Encoding a Tumor-Specific Chimeric Antigen Receptor. Cancer Immunol. Immunother. CII 2014, 63, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial Antigen Recognition with Balanced Signaling Promotes Selective Tumor Eradication by Engineered T Cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific Antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, J.; Zhong, R.; Mokotoff, M.; Shultz, L.D.; Ball, E.D. Targeting Gastrin-Releasing Peptide Receptors on Small Cell Lung Cancer Cells with a Bispecific Molecule That Activates Polyclonal T Lymphocytes. Clin. Cancer Res. 2006, 12, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Völger, H.R.; Lorenz, S.; Imhof-Jung, S.; Regula, J.T.; Klein, C.; Mølhøj, M. Heavy and Light Chain Pairing of Bivalent Quadroma and Knobs-into-Holes Antibodies Analyzed by UHR-ESI-QTOF Mass Spectrometry. MAbs 2015, 8, 49–55. [Google Scholar] [CrossRef]
- Ridgway, J.B.B.; Presta, L.G.; Carter, P. Knobs-into-Holes Engineering of Antibody CH3 Domains for Heavy Chain Heterodimerization. Protein Eng. Des. Sel. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T Cell Engagers for Cancer Immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Yolken, R.H.; Peterson, J.A.; Vonderfecht, S.L.; Fouts, E.T.; Midthun, K.; Newburg, D.S. Human Milk Mucin Inhibits Rotavirus Replication and Prevents Experimental Gastroenteritis. J. Clin. Invest. 1992, 90, 1984–1991. [Google Scholar] [CrossRef]
- Schroten, H.; Hanisch, F.G.; Plogmann, R.; Hacker, J.; Uhlenbruck, G.; Nobis-Bosch, R.; Wahn, V. Inhibition of Adhesion of S-Fimbriated Escherichia Coli to Buccal Epithelial Cells by Human Milk Fat Globule Membrane Components: A Novel Aspect of the Protective Function of Mucins in the Nonimmunoglobulin Fraction. Infect. Immun. 1992, 60, 2893–2899. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. Muc1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Dalziel, M.; Whitehouse, C.; McFarlane, I.; Brockhausen, I.; Gschmeissner, S.; Schwientek, T.; Clausen, H.; Burchell, J.M.; Taylor-Papadimitriou, J. The Relative Activities of the C2GnT1 and ST3Gal-I Glycosyltransferases Determine O-Glycan Structure and Expression of a Tumor-Associated Epitope on MUC1. J. Biol. Chem. 2001, 276, 11007–11015. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.D.; Dillon, L.M.; Zhou, R.; Moore, L.J.; Livasy, C.; El-Khoury, J.M.; Puri, R.; Mukherjee, P. A Tumor Specific Antibody to Aid Breast Cancer Screening in Women with Dense Breast Tissue. Genes Cancer 2017, 8, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Liang, H.; Hao, C.; Yang, X.; Cui, X. Overexpression of MUC1 Predicts Poor Prognosis in Patients with Breast Cancer. Oncol. Rep. 2019, 41, 801–810. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Wallstabe, L.; Mades, A.; Frenz, S.; Einsele, H.; Rader, C.; Hudecek, M. CAR T Cells Targeting Avβ3 Integrin Are Effective against Advanced Cancer in Preclinical Models. Adv. Cell Gene Ther. 2018, 1. [Google Scholar] [CrossRef]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Genetically Modified T Cells Targeting Neovasculature Efficiently Destroy Tumor Blood Vessels, Shrink Established Solid Tumors, and Increase Nanoparticle Delivery. Int. J. Cancer 2013, 133, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Gutheil, J.C.; Campbell, T.N.; Pierce, P.R.; Watkins, J.D.; Huse, W.D.; Bodkin, D.J.; Cheresh, D.A. Targeted Antiangiogenic Therapy for Cancer Using Vitaxin: A Humanized Monoclonal Antibody to the Integrin Alphavbeta3. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2000, 6, 3056–3061. [Google Scholar]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A Randomized Phase 2 Study of Etaracizumab, a Monoclonal Antibody against Integrin Alpha(v) Beta (3), + or—Dacarbazine in Patients with Stage IV Metastatic Melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Carson-Walter, E.B.; Watkins, D.N.; Nanda, A.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. Cell Surface Tumor Endothelial Markers Are Conserved in Mice and Humans. Cancer Res. 2001, 61, 6649–6655. [Google Scholar] [PubMed]
- Chaudhary, A.; Hilton, M.B.; Seaman, S.; Haines, D.C.; Stevenson, S.; Lemotte, P.K.; Tschantz, W.R.; Zhang, X.M.; Saha, S.; Fleming, T.; et al. TEM8/ANTXR1 Blockade Inhibits Pathological Angiogenesis and Potentiates Tumoricidal Responses against Multiple Cancer Types. Cancer Cell 2012, 21, 212–226. [Google Scholar] [CrossRef]
- Croix, B.S.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes Expressed in Human Tumor Endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef]
- Vargas, M.; Karamsetty, R.; Leppla, S.H.; Chaudry, G.J. Broad Expression Analysis of Human ANTXR1/TEM8 Transcripts Reveals Differential Expression and Novel Splizce Variants. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Davies, G.; Rmali, K.A.; Watkins, G.; Mansel, R.E.; Mason, M.D.; Jiang, W.G. Elevated Levels of Tumour Endothelial Marker-8 in Human Breast Cancer and Its Clinical Significance. Int. J. Oncol. 2006, 29, 1311–1317. [Google Scholar] [CrossRef]
- Gutwein, L.G.; Al-Quran, S.Z.; Fernando, S.; Fletcher, B.S.; Copeland, E.M.; Grobmyer, S.R. Tumor Endothelial Marker 8 Expression in Triple-Negative Breast Cancer. Anticancer Res. 2011, 31, 3417–3422. [Google Scholar]
- Byrd, T.T.; Fousek, K.; Pignata, A.; Szot, C.; Samaha, H.; Seaman, S.; Dobrolecki, L.; Salsman, V.S.; Oo, H.Z.; Bielamowicz, K.; et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Cui, B.; Chuang, H.-Y.; Yu, J.; Wang-Rodriguez, J.; Tang, L.; Chen, G.; Basak, G.W.; Kipps, T.J. ROR1 is Expressed in Human Breast Cancer and Associated with Enhanced Tumor-Cell Growth. PLoS ONE 2012, 7, e31127. [Google Scholar] [CrossRef] [PubMed]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T Cells Are Effective against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.-E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor Efficacy of a Bispecific Antibody That Targets HER2 and Activates T Cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef]
- Ichnos Sciences, S.A. A Phase 1/2, Open-Label, Dose-Escalation Study of ISB 1302 in Subjects with HER2-Positive Metastatic Breast Cancer; ClinicalTrials.gov: Bethesda, MD, USA, 2020.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Gutierrez, C.; Schiff, R. HER 2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef]
- Kallioniemi, O.P.; Kallioniemi, A.; Kurisu, W.; Thor, A.; Chen, L.C.; Smith, H.S.; Waldman, F.M.; Pinkel, D.; Gray, J.W. ERBB2 Amplification in Breast Cancer Analyzed by Fluorescence in Situ Hybridization. Proc. Natl. Acad. Sci. USA 1992, 89, 5321–5325. [Google Scholar] [CrossRef]
- Ruiz, I.R.; Vicario, R.; Morancho, B.; Morales, C.B.; Arenas, E.J.; Herter, S.; Freimoser-Grundschober, A.; Somandin, J.; Sam, J.; Ast, O.; et al. P95HER2–T Cell Bispecific Antibody for Breast Cancer Treatment. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hooper, A.T.; Zhong, Z.; Witte, L.; Bohlen, P.; Rafii, S.; Hicklin, D.J. The Vascular Endothelial Growth Factor Receptor (VEGFR-1) Supports Growth and Survival of Human Breast Carcinoma. Int. J. Cancer 2006, 119, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Li, L.; Zhou, Y.; Shen, C.-C.; Kang, Y.-H.; Yao, Y.-Q.; Yi, C.; Gou, L.-T.; Yang, J.-L. The Preparation of VEGFR1/CD3 Bispecific Antibody and Its Specific Cytotoxicity against VEGFR1-Positive Breast Cancer Cells. Biotechnol. Appl. Biochem. 2014, 61, 376–384. [Google Scholar] [CrossRef]
- Al-Chalabi, M.; Bass, A.N.; Alsalman, I. Physiology, Prolactin; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Zhou, Y.; Zong, H.; Han, L.; Xie, Y.; Jiang, H.; Gilly, J.; Zhang, B.; Lu, H.; Chen, J.; Sun, R.; et al. A Novel Bispecific Antibody Targeting CD3 and Prolactin Receptor (PRLR) against PRLR-Expression Breast Cancer. J. Exp. Clin. Cancer Res. CR 2020, 39. [Google Scholar] [CrossRef]
- Andreev, J. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L. Molecular Mechanisms of Prolactin and Its Receptor. Endocr. Rev. 2012, 33, 504–525. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Machiels, J.-P.; Suárez, C.; Lewis, N.; Higgins, M.; Wisinski, K.; Awada, A.; Maur, M.; Stein, M.; Hwang, A.; et al. Phase I Study of the Prolactin Receptor Antagonist LFA102 in Metastatic Breast and Castration-Resistant Prostate Cancer. Oncologist 2016, 21, 535–536. [Google Scholar] [CrossRef]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396. [Google Scholar] [CrossRef]
- Necela, B.M.; Crozier, J.A.; Andorfer, C.A.; Lewis-Tuffin, L.; Kachergus, J.M.; Geiger, X.J.; Kalari, K.R.; Serie, D.J.; Sun, Z.; Aspita, A.M.; et al. Folate Receptor-α (FOLR1) Expression and Function in Triple Negative Tumors. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Lv, Q.; Yang, J.; Zhang, R.; Yang, Z.; Yang, Z.; Wang, Y.; Xu, Y.; He, Z. Prostate-Specific Membrane Antigen Targeted Therapy of Prostate Cancer Using a DUPA-Paclitaxel Conjugate. Mol. Pharm. 2018, 15, 1842–1852. [Google Scholar] [CrossRef]
- Pastorekova, S.; Gillies, R.J. The Role of Carbonic Anhydrase IX in Cancer Development: Links to Hypoxia, Acidosis, and Beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef]
- O’Keefe, D.S.; Bacich, D.J.; Huang, S.S.; Heston, W.D.W. A Perspective on the Evolving Story of PSMA Biology, PSMA-Based Imaging, and Endoradiotherapeutic Strategies. J. Nucl. Med. 2018, 59, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Svastová, E.; Hulíková, A.; Rafajová, M.; Zat’ovicová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia Activates the Capacity of Tumor-Associated Carbonic Anhydrase IX to Acidify Extracellular PH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef]
- Pastorek, J.; Pastoreková, S.; Callebaut, I.; Mornon, J.P.; Zelník, V.; Opavský, R.; Zat’ovicová, M.; Liao, S.; Portetelle, D.; Stanbridge, E.J. Cloning and Characterization of MN, a Human Tumor-Associated Protein with a Domain Homologous to Carbonic Anhydrase and a Putative Helix-Loop-Helix DNA Binding Segment. Oncogene 1994, 9, 2877–2888. [Google Scholar]
- Horie, K.; Kawakami, K.; Fujita, Y.; Sugaya, M.; Kameyama, K.; Mizutani, K.; Deguchi, T.; Ito, M. Exosomes Expressing Carbonic Anhydrase 9 Promote Angiogenesis. Biochem. Biophys. Res. Commun. 2017, 492, 356–361. [Google Scholar] [CrossRef]
- PubChem Acetazolamide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/1986 (accessed on 22 December 2020).
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Deng, G. Tumor-Infiltrating Regulatory T Cells: Origins and Features. Am. J. Clin. Exp. Immunol. 2018, 7, 81–87. [Google Scholar]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 Coreceptor Expression and Signal Transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.; Fouser, L.A.; Jussif, J.; Fitz, L.; Deng, B.; Wood, C.R.; Collins, M.; Honjo, T.; Freeman, G.J.; Carreno, B.M. PD-1: PD-L Inhibitory Pathway Affects Both CD4(+) and CD8(+) T Cells and Is Overcome by IL-2. Eur. J. Immunol. 2002, 32, 634–643. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective Effects of PD–1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation. Sci. Signal. 2012, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP–1 and SHP–2 Associate with Immunoreceptor Tyrosine–Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Nurieva, R.; Thomas, S.; Nguyen, T.; Martin–Orozco, N.; Wang, Y.; Kaja, M. –K.; Yu, X.–Z.; Dong, C. T–Cell Tolerance or Function Is Determined by Combinatorial Costimulatory Signals. EMBO J. 2006, 25, 2623–2633. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T Cell Co–Receptors for Cancer Therapy. Immunity 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo Imaging Reveals a Tumor—Associated Macrophage—Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Miao, L.; Liu, Q.; Musetti, S.; Li, J.; Huang, L. Combination Immunotherapy of MUC1 MRNA Nano-Vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol. Ther. 2018, 26, 45–55. [Google Scholar] [CrossRef]
- Kaur, S.P.; Gupta, V. COVID-19 Vaccine: A Comprehensive Status Report. Virus Res. 2020, 288, 198114. [Google Scholar] [CrossRef]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The New Revolution in Nucleic Acid Vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Wessman, D. MRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and Efficacy of Combination Treatment with Anti-LAG-3 and Anti-PD-1 Monoclonal Antibodies in Glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; Marzo, A.D.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 Regulates CD8+ T Cell Accumulation and Effector Function in Murine Self and Tumor-Tolerance Systems. J. Clin. Invest. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and Its Role in Regulating Anti-Tumor Immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.-W.; Dutta, A.; Chang, L.-Y.; Mahalingam, J.; Lin, Y.-C.; Chiang, J.-M.; Hsu, C.-Y.; Huang, C.-T.; Su, W.-T.; Chu, Y.-Y.; et al. Apoptosis of Tumor Infiltrating Effector TIM-3+CD8+ T Cells in Colon Cancer. Sci. Rep. 2015, 5, 15659. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; Scheidt, B.; Von Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 Antibody Promotes T Cell IFN-γ–Mediated Antitumor Immunity and Suppresses Established Tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Stamm, H.; Oliveira-Ferrer, L.; Grossjohann, E.-M.; Muschhammer, J.; Thaden, V.; Brauneck, F.; Kischel, R.; Müller, V.; Bokemeyer, C.; Fiedler, W.; et al. Targeting the TIGIT-PVR Immune Checkpoint Axis as Novel Therapeutic Option in Breast Cancer. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhao, W.; Li, H.; Chen, Y.; Tian, H.; Li, L.; Zhang, L.; Gao, C.; Zheng, J. Immunoreceptor TIGIT Inhibits the Cytotoxicity of Human Cytokine-Induced Killer Cells by Interacting with CD155. Cancer Immunol. Immunother. CII 2016, 65, 305–314. [Google Scholar] [CrossRef]
- Eccles, S.A. The Epidermal Growth Factor Receptor/Erb-B/HER Family in Normal and Malignant Breast Biology. Int. J. Dev. Biol. 2011, 55, 685–696. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of Epidermal Growth Factor Receptor in Breast Cancer. Breast Cancer Res. Treat. 2012, 136. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, F.; Fry, T.J.; Milliron, M.J.; McKirdy, M.A.; Tagaya, Y.; Mackall, C.L. Adjuvant IL-7 or IL-15 Overcomes Immunodominance and Improves Survival of the CD8+ Memory Cell Pool. J. Clin. Invest. 2005, 115, 1177–1187. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res. Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Colavito, S.A. AXL as a Target in Breast Cancer Therapy. J. Oncol. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-Receptor Signaling Augments the Antitumor Function of ROR1-Specific CAR T-Cells against Triple-Negative Breast Cancer. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Target | Cellular/Molecular Function | Tumor Specific (TS) /Tumor Associated (TA) | Malignant Transformation |
|---|---|---|---|
| MUC1 (glycosylated transmembrane protein); tMUC1 (mutated) | Produces mucin; protective barrier against pathogens | TS | Over-expressed and hypoglycosylated; exposure of new protein epitopes |
| αvβ3 integrin | Promotes migration, invasion, survival, and proliferation | TA | Overexpressed |
| TEM8 (tumor endothelial marker), ANTXR1 | Endothelial cell development | TA | Overexpressed |
| ROR1 (receptor tyrosine kinase-like orphan receptor 1) | Highly expressed during embryogenesis; promotes migration, invasion, anti-apoptosis | TA | Overexpressed |
| Mesothelin (glycosylphosphatidylinositol- anchored cell surface receptor) | Migration and invasion | TA | Ove-expressed |
| AXL (receptor tyrosine kinase) | Cell growth and survival | TA | Overexpressed |
| Molecular Target | Cellular/Molecular Function | Tumor Specific (TS)/Tumor Associated (TA) | Malignant Transformation |
|---|---|---|---|
| HER2 (transmembrane glycoprotein receptor; epidermal growth factor receptor (EGFR) family); p95HER2 (mutated) | Survival and migration | TS | Truncated version of HER2 only expressed in HER2+ breast cancer |
| VEGFR1 (vascular endothelial growth factor receptor 1) | Receptor tyrosine kinase; Migration, survival, proliferation | TA | Overexpressed |
| PRLR (prolactin receptor) | Milk production and mammary gland development | TA | Overexpressed |
| FRα receptor (folate receptor) | Transportation of folate; DNA synthesis biochemical pathway | TA | Overexpressed; BsAb cocktail therapy |
| EGFR (transmembrane glycoprotein) | Proliferation, adhesion, anti-apoptosis, invasion, angiogenesis | TA | Overexpressed |
| Immune Checkpoint/T Cell Signal | Function | Therapy Explored in Review |
|---|---|---|
| PD-1 (programmed cell death protein 1) | Reduction in pro-survival gene expression within the BCL family, cytokine production, cell cycle progression | CRISPR-CAS9 knockout of PD-1 in CAR-T cells against mesothelin in TNBC |
| CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) | Inhibit costimulatory signal, inhibit T cell activation by competitively binding CD80 or CD86 | mRNA vaccine against MUC1 in TNBC combined with anti-CTLA-4 antibody |
| LAG-3 (lymphocyte-activation protein 3) | Apoptosis, decreased proliferation of T cells | anti-LAG-3 antibody |
| TIM-3 (T cell immunoglobulin and mucin domain-containing protein 3) | Reduces immune response in chronic inflammation and tumor infiltrating leukocytes | anti-TIM-3 antibody |
| TIGIT (T cell immunoreceptor with immunoglobulin and ITIM domains) | Reduces T cell proliferation and cytokine production | BsAb therapy against EGFR in combination with anti-TIGIT-antibody |
| IL-7R (interleukin 7 receptor) | Proliferation, anti-apoptosis of T cells | Constitutively active IL-7R modified CAR-T cells against AXL |
| TGF-β (transforming growth factor beta) | Inhibits general functions of immune cell | CAR-T therapy against ROR1 in combination with TGF-β inhibitor (SD-208) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivaganesh, V.; Promi, N.; Maher, S.; Peethambaran, B. Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 2433. https://doi.org/10.3390/ijms22052433
Sivaganesh V, Promi N, Maher S, Peethambaran B. Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer. International Journal of Molecular Sciences. 2021; 22(5):2433. https://doi.org/10.3390/ijms22052433
Chicago/Turabian StyleSivaganesh, Vignesh, Nazifa Promi, Salma Maher, and Bela Peethambaran. 2021. "Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer" International Journal of Molecular Sciences 22, no. 5: 2433. https://doi.org/10.3390/ijms22052433
APA StyleSivaganesh, V., Promi, N., Maher, S., & Peethambaran, B. (2021). Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer. International Journal of Molecular Sciences, 22(5), 2433. https://doi.org/10.3390/ijms22052433