
Comparative Genomics: Insights on the Pathogenicity and Lifestyle of Rhizoctonia solani

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Genome Annotation of Draft AG1-IA & Comparative Genome Assembly Statistics
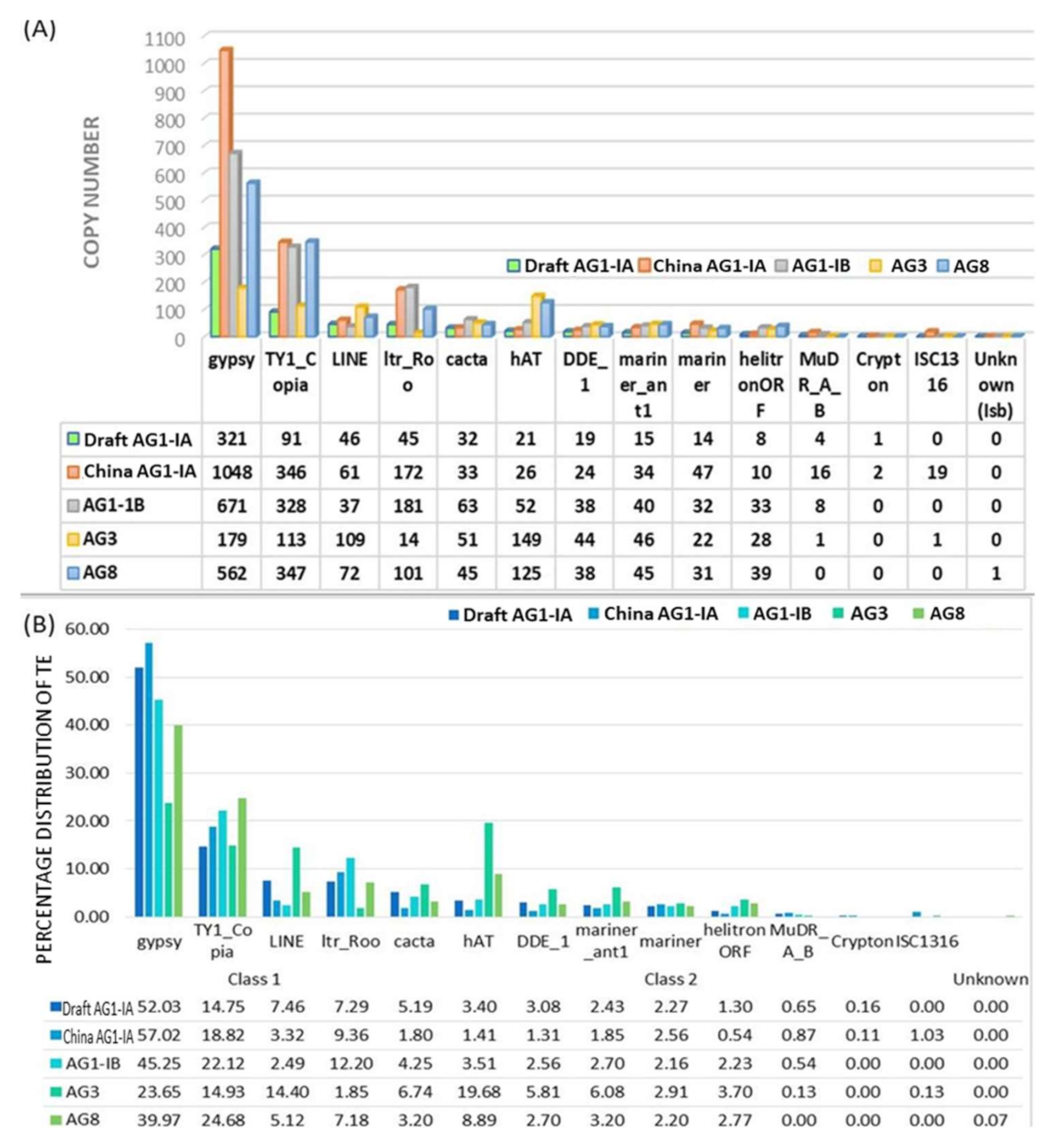
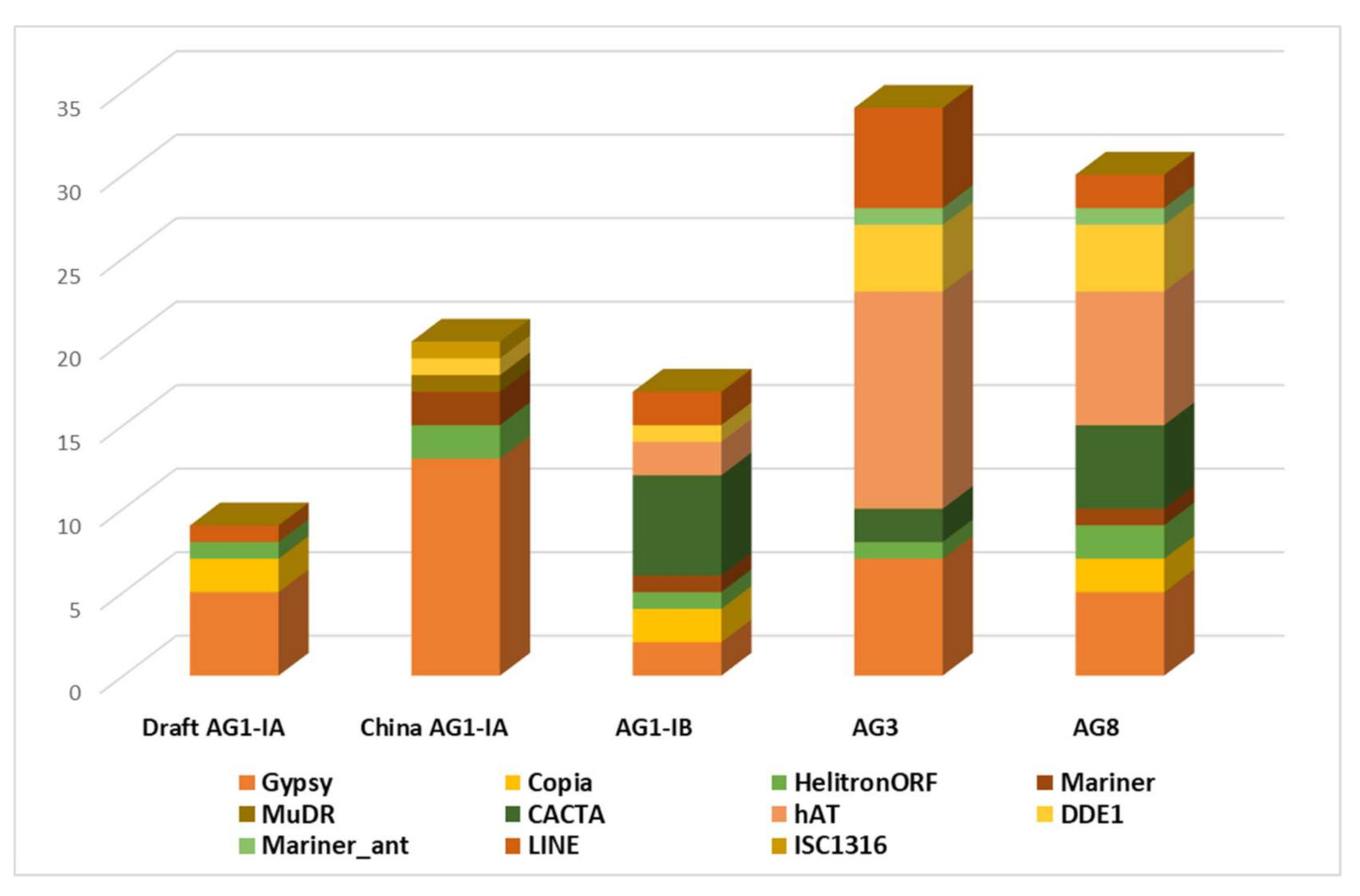
2.2. Repeat Element Identification and Characterization of Transposable Element
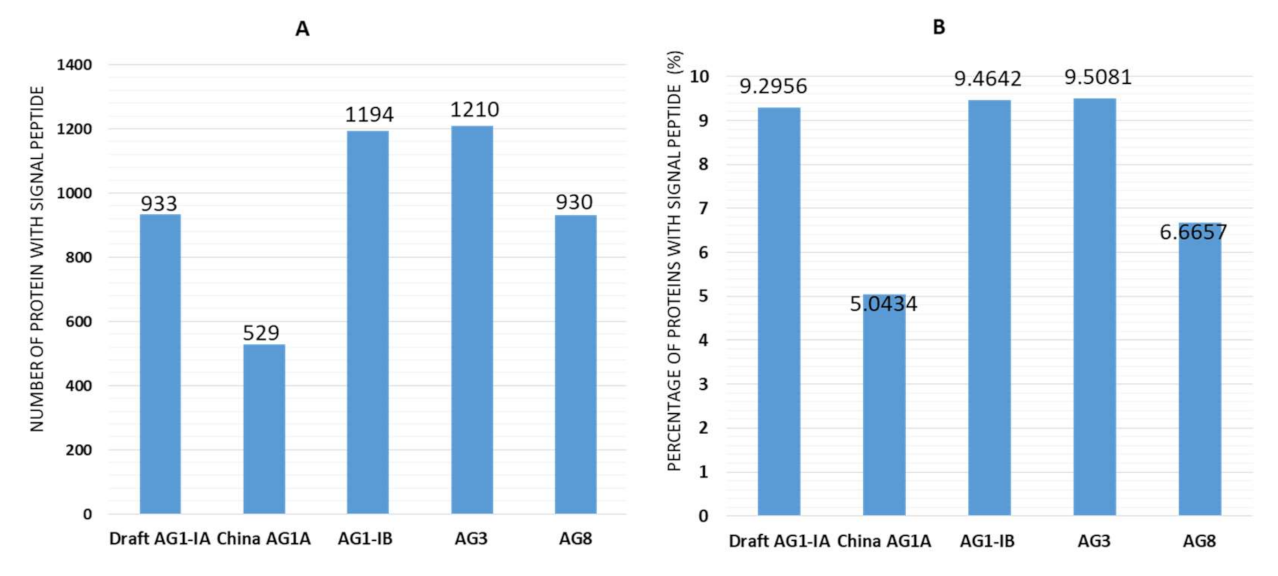
2.3. Signal Peptides
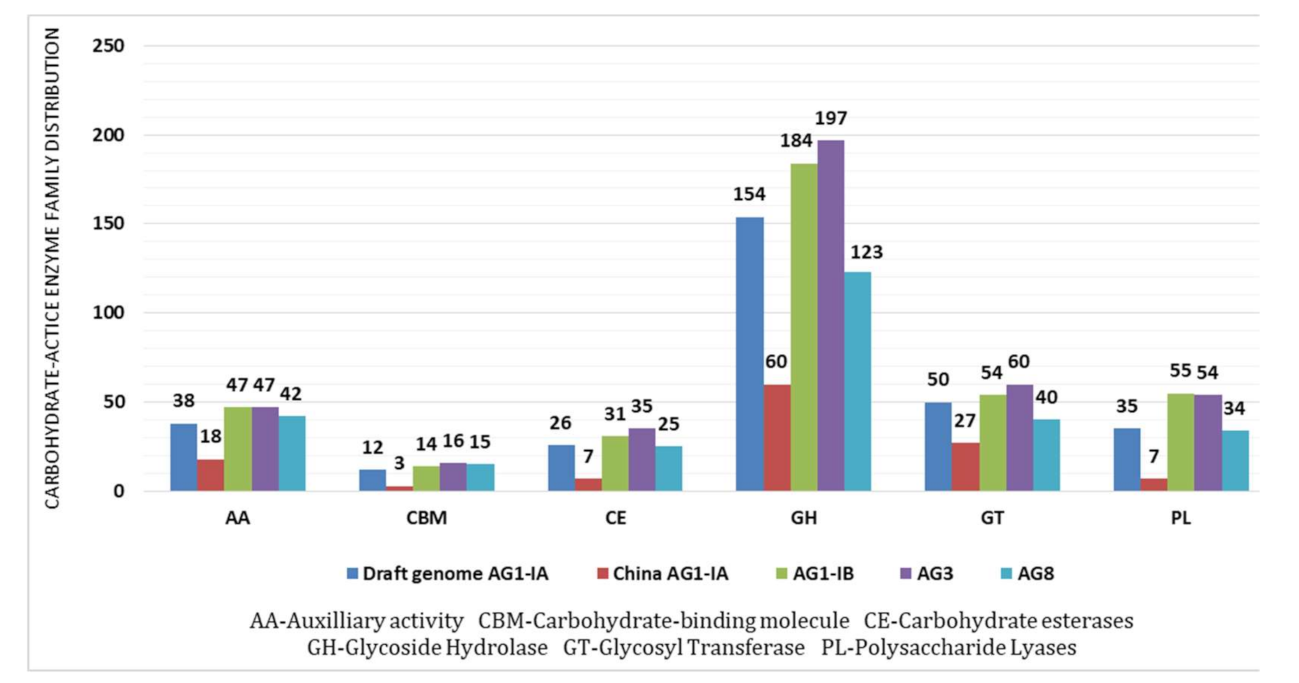
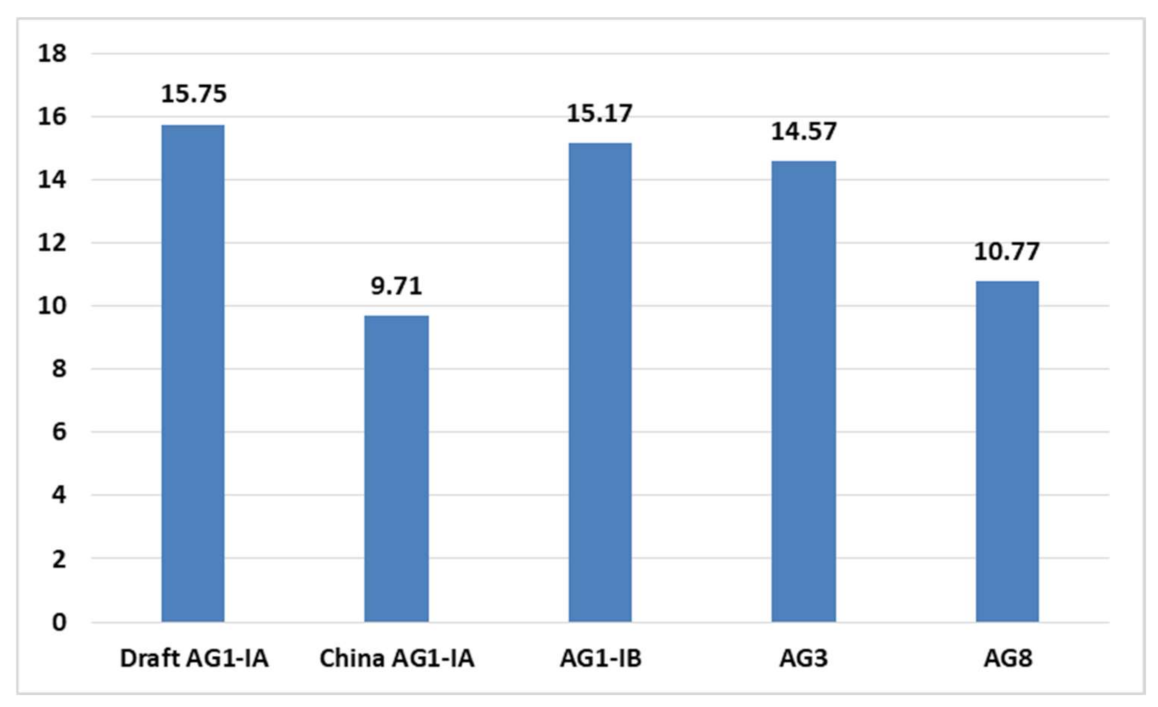
2.4. CAZy Distribution Analysis
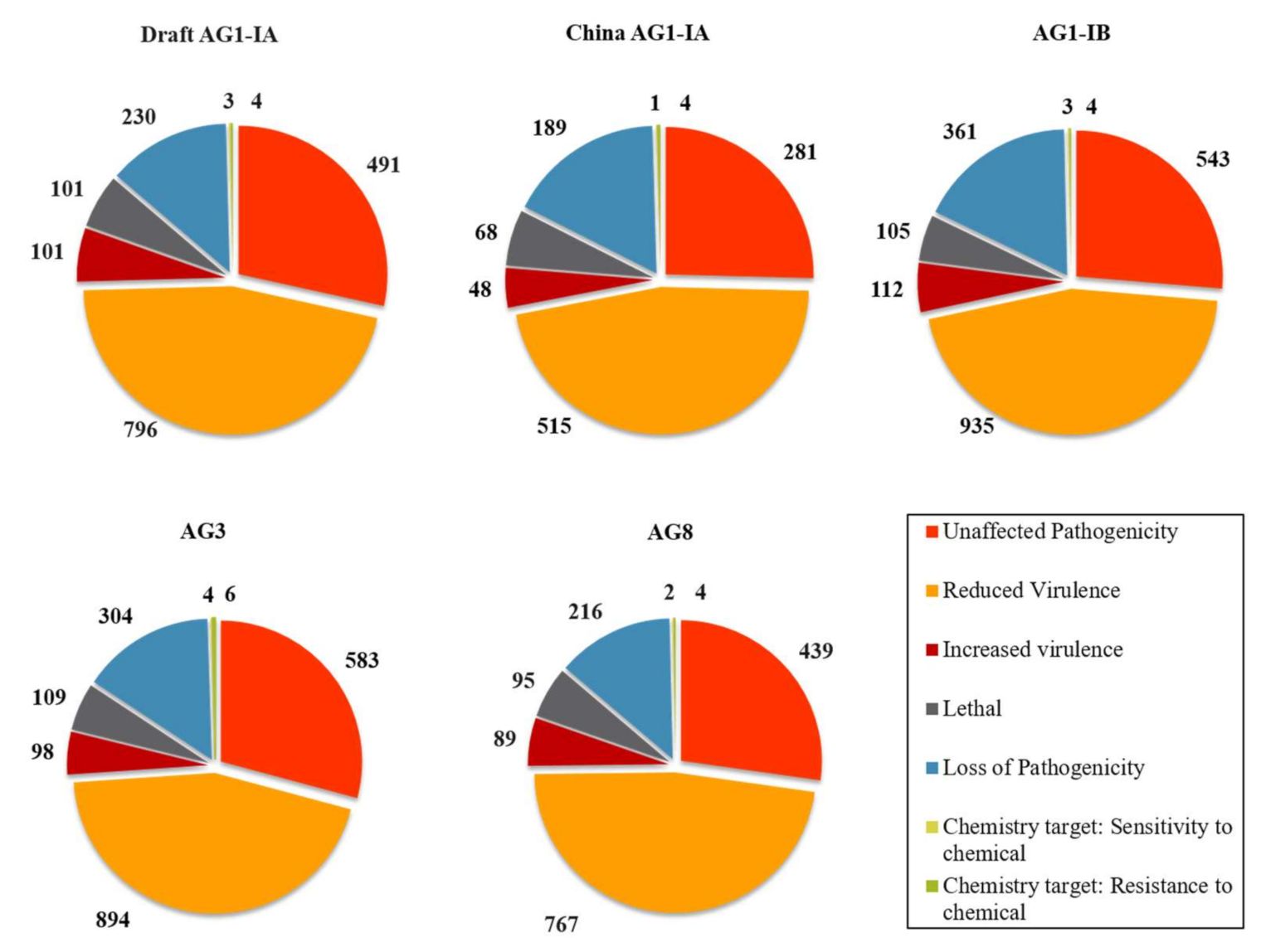
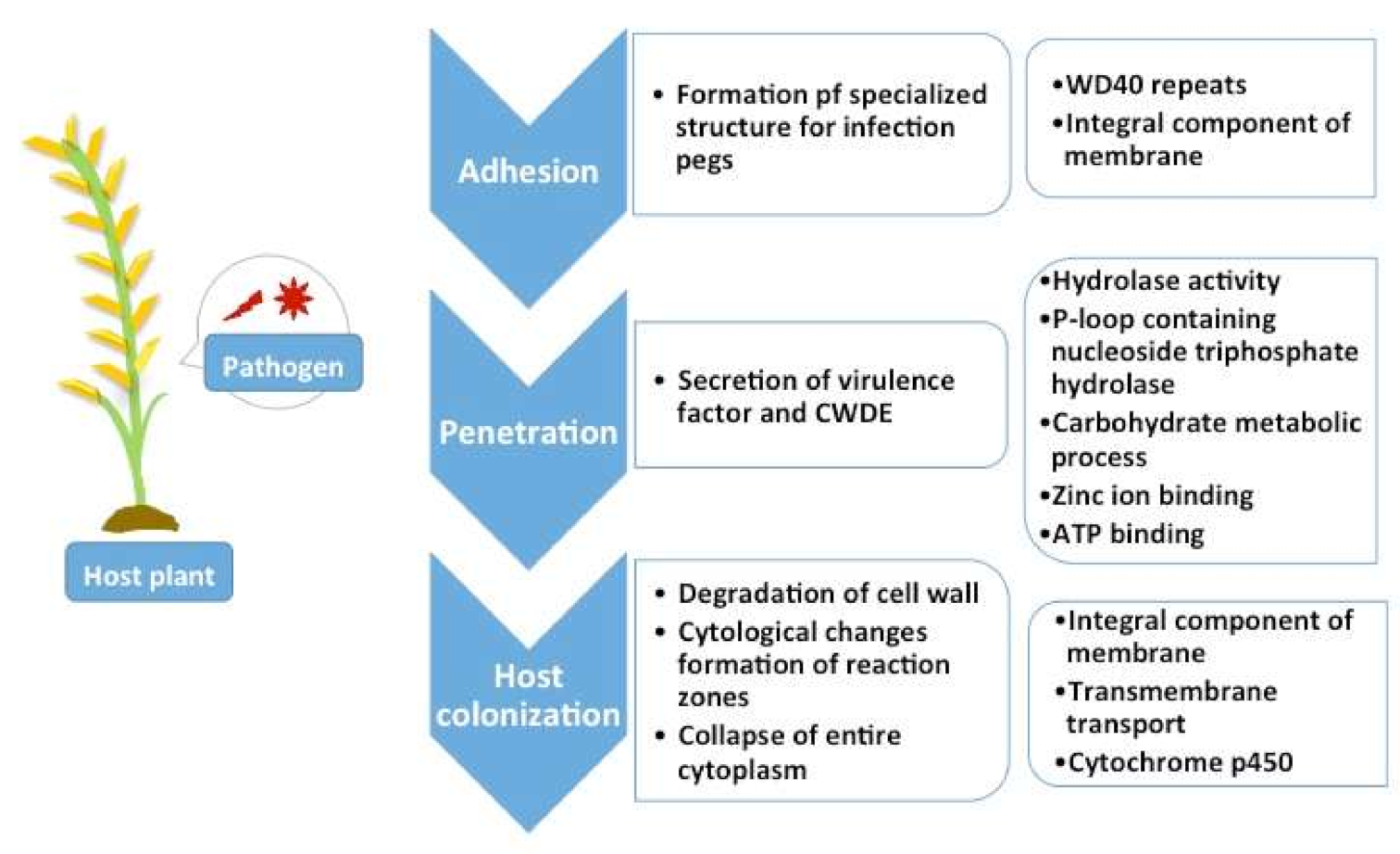
2.5. Pathogenicity Genes Distribution and the Impact of TEs on Pathogenicity
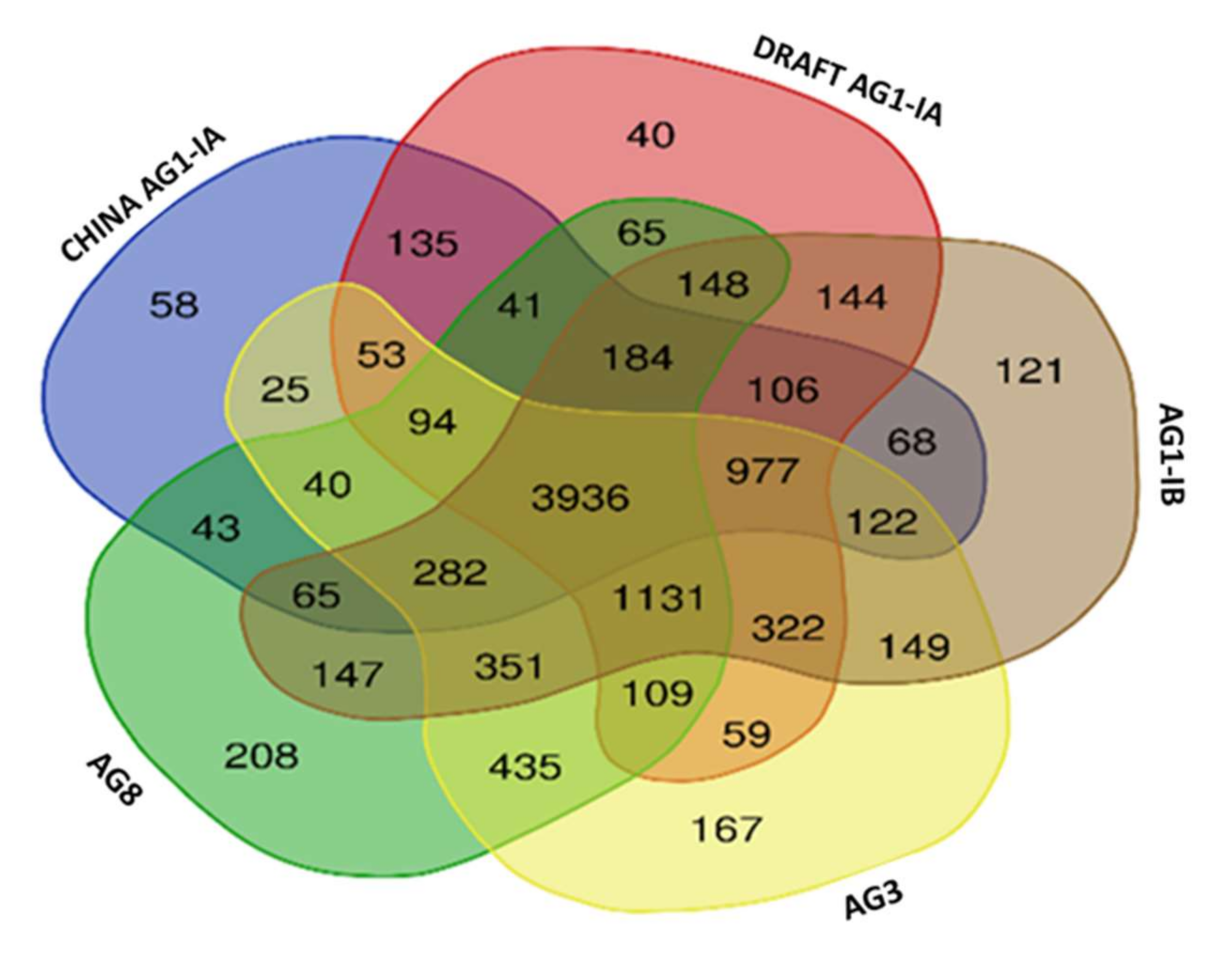
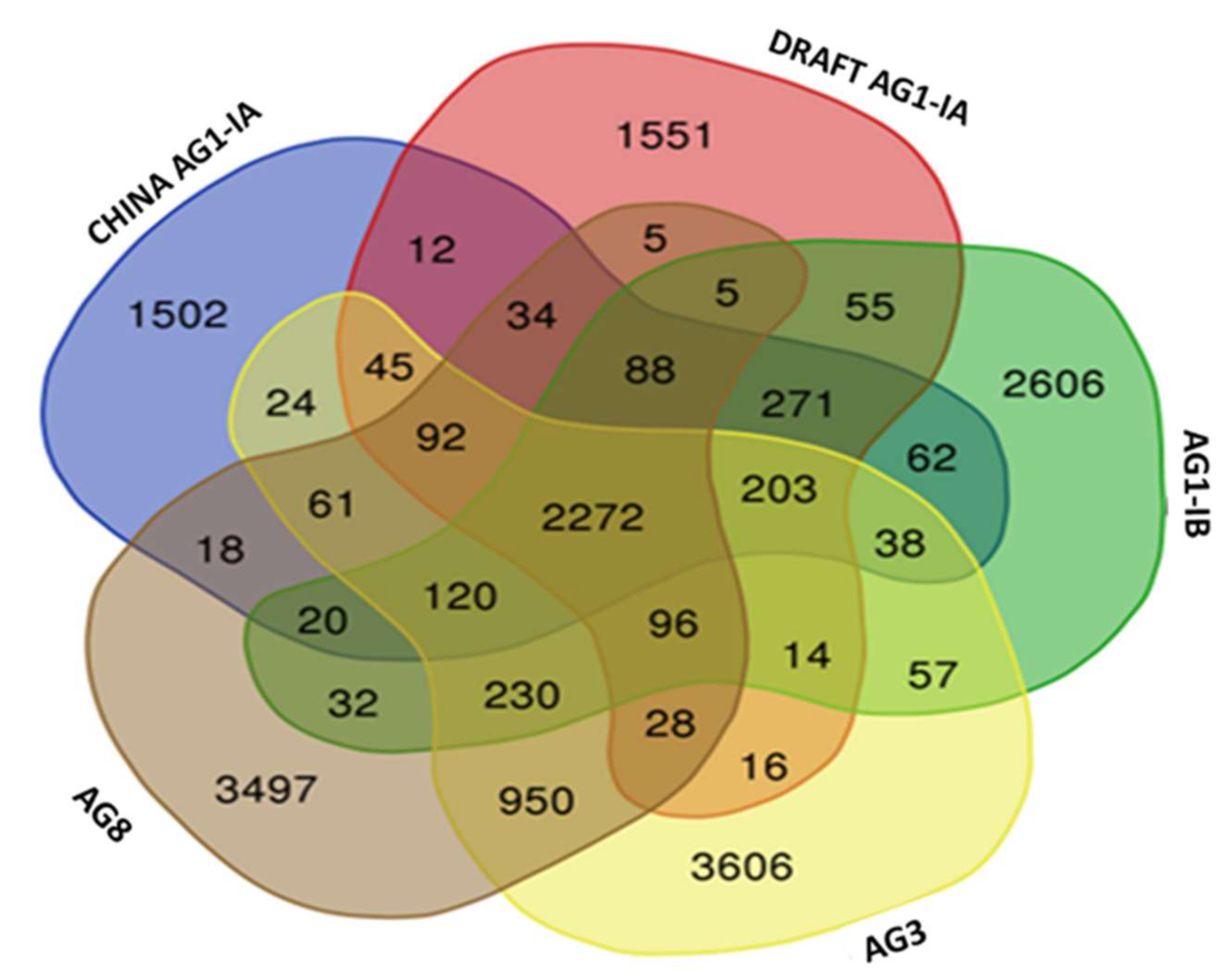
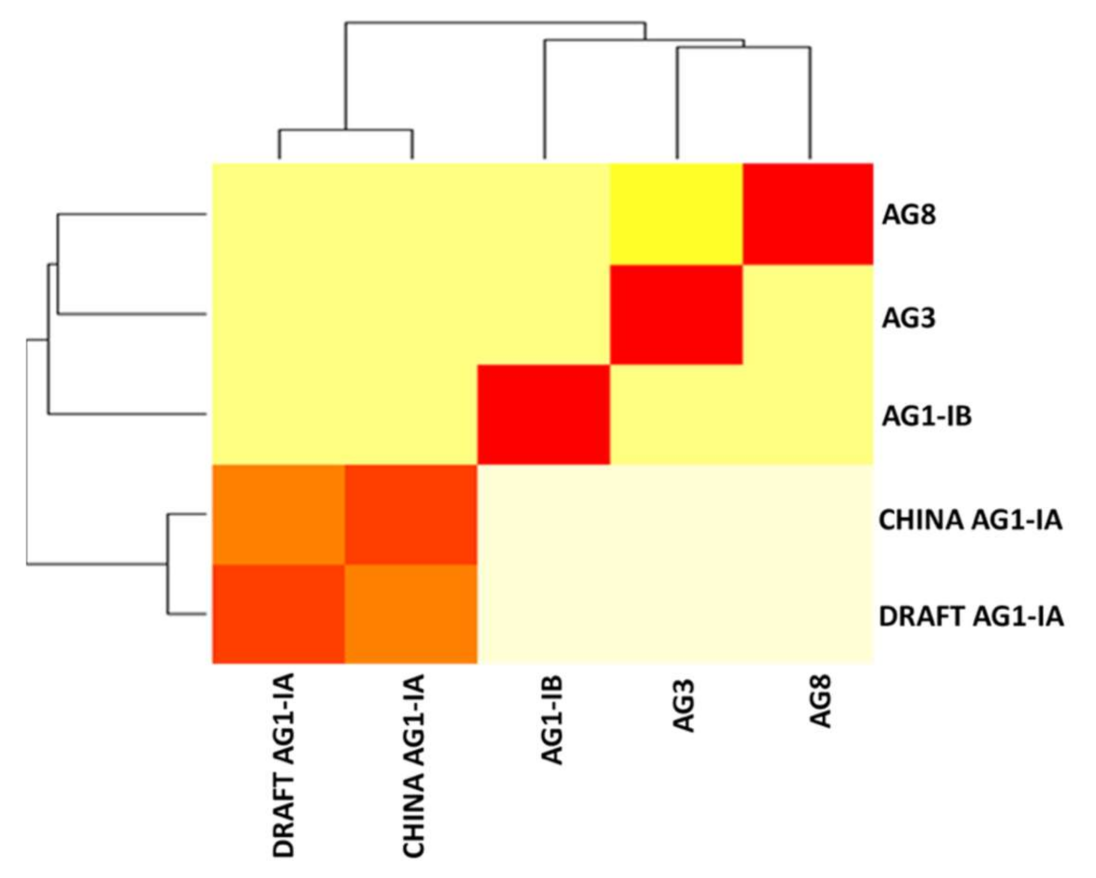
2.6. Single Copy Orthologs
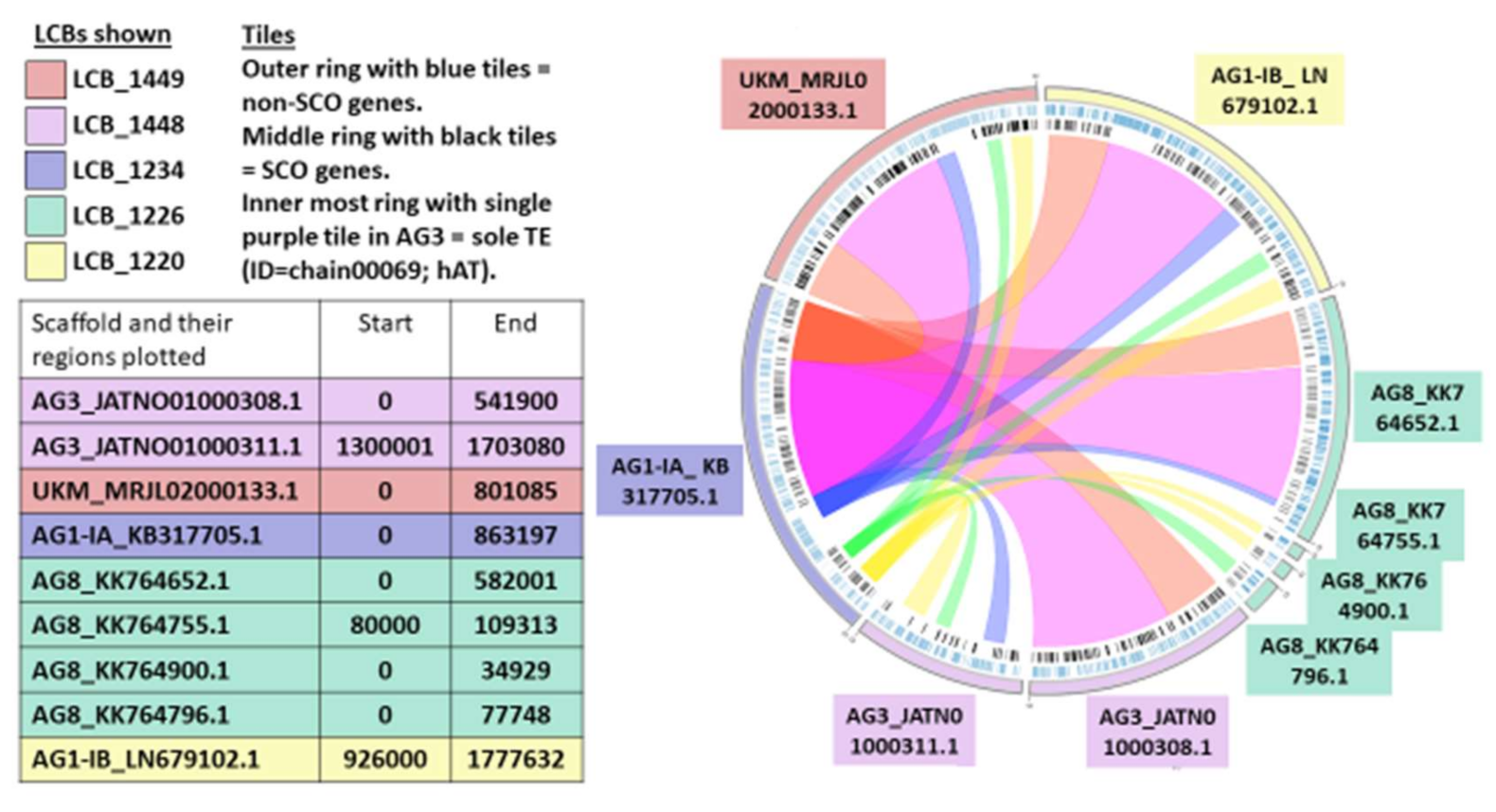
2.7. Synteny Analysis
3. Materials and Methods
3.1. Gene prediction & Genome Annotation
3.2. Repeat Elements Characterization
3.3. TE Identification
3.4. Pathogenicity Gene Distribution
3.5. Proximity Analysis: Link to TE Influence in Pathogenicity
3.6. Signal Peptides
3.7. Carbohydrate Active Enzymes (CAZy)
- CAZyDB.07202017.fa.
- CAZyDB-ec-info.txt.
- CAZyDB-ec-info.txt.07-20-2017.
3.8. Single Copy Ortholog Analysis
3.9. Synteny Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagaraj, B.; Sunkad, G.; Pramesh, D.; Naik, M.; Patil, M. Host range studies of rice sheath blight fungus Rhizoctonia solani (Kuhn). Int. J. Curr. Microbiol. App. Sci 2017, 6, 3856–3864. [Google Scholar] [CrossRef]
- García, V.G.; Onco, M.P.; Susan, V.R. Biology and systematics of the form genus Rhizoctonia. Span. J. Agric. Res. 2006, 4, 55–79. [Google Scholar] [CrossRef]
- Wibberg, D.; Jelonek, L.; Rupp, O.; Hennig, M.; Eikmeyer, F.; Goesmann, A.; Hartmann, A.; Borriss, R.; Grosch, R.; Pühler, A. Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J. Biotechnol. 2013, 167, 142–155. [Google Scholar] [CrossRef]
- Loan, L.; Du, P.; Li, Z. Molecular dissection of quantitative resistance of sheath blight in rice (Oryza sativa L.). Omonrice 2004, 12, 1–12. [Google Scholar]
- Pinson, S.R.; Capdevielle, F.M.; Oard, J.H. Confirming QTLs and finding additional loci conditioning sheath blight resistance in rice using recombinant inbred lines. Crop Sci. 2005, 45, 503–510. [Google Scholar] [CrossRef]
- Liu, S.; Hall, M.D.; Griffey, C.A.; McKendry, A.L. Meta-analysis of QTL associated with Fusarium head blight resistance in wheat. Crop Sci. 2009, 49, 1955–1968. [Google Scholar] [CrossRef]
- Jia, L.; Yan, W.; Zhu, C.; Agrama, H.A.; Jackson, A.; Yeater, K.; Li, X.; Huang, B.; Hu, B.; McClung, A. Allelic analysis of sheath blight resistance with association mapping in rice. PLoS ONE 2012, 7, e32703. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Maurer, H.; Reif, J.; Melchinger, A.; Utz, H.; Tucker, M.; Ranc, N.; Della Porta, G.; Würschum, T. Optimum design of family structure and allocation of resources in association mapping with lines from multiple crosses. Heredity 2013, 110, 71. [Google Scholar] [CrossRef]
- Kumar, I.S.; Cheah, B.; Nadarajah, K. In silico identification and classification of disease resistance genes and defense-related genes against sheath blight from QTL qSBR11-1 in rice (Oryza sativa L.). Undergrad. Res. J. Integ. Biol. 2017, 1, 8. [Google Scholar]
- Kumar, I.S.; Zaharin, N.; Nadarajah, K. In silico Identification of Resistance and Defense Related Genes for Bacterial Leaf Blight (BLB) in Rice. J. Pure Appl. Microbiol. 2018, 12, 1867–1877. [Google Scholar] [CrossRef]
- Kumar, I.S.; Amran, N.A.; Nadarajah, K. In silico Analysis of qBFR4 and qLBL5 in Conferring Quantitative Resistance Against Rice Blast. J. Pure Appl. Microbiol. 2018, 12, 1703–1718. [Google Scholar] [CrossRef]
- Chanthran, S.S.D.; Cheah, B.H.; Nadarajah, K.K. In silico analysis of disease resistance and defence-related genes for a major sheath blight qShb 9-2 QTL in rice. Malays. J. Microbiol. 2018, 14, 534–546. [Google Scholar] [CrossRef]
- Walton, J.D. Host-selective toxins: Agents of compatibility. Plant Cell 1996, 8, 1723. [Google Scholar]
- Friesen, T.L.; Zhang, Z.; Solomon, P.S.; Oliver, R.P.; Faris, J.D. Characterization of the interaction of a novel Stagonospora nodorum host-selective toxin with a wheat susceptibility gene. Plant Physiol. 2008, 146, 682–693. [Google Scholar] [CrossRef]
- Laluk, K.; Mengiste, T. Necrotroph attacks on plants: Wanton destruction or covert extortion? Arab. Book/Am. Soc. Plant Biol. 2010, 8. [Google Scholar] [CrossRef]
- Verwaaijen, B.; Wibberg, D.; Kröber, M.; Winkler, A.; Zrenner, R.; Bednarz, H.; Niehaus, K.; Grosch, R.; Pühler, A.; Schlüter, A. The Rhizoctonia solani AG1-IB (isolate 7/3/14) transcriptome during interaction with the host plant lettuce (Lactuca sativa L.). PLoS ONE 2017, 12, e0177278. [Google Scholar] [CrossRef]
- Ghosh, S.; Kanwar, P.; Jha, G. Identification of candidate pathogenicity determinants of Rhizoctonia solani AG1-IA, which causes sheath blight disease in rice. Curr. Genet. 2018, 64, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Cubeta, M.A.; Thomas, E.; Dean, R.A.; Jabaji, S.; Neate, S.M.; Tavantzis, S.; Toda, T.; Vilgalys, R.; Bharathan, N.; Fedorova-Abrams, N. Draft genome sequence of the plant-pathogenic soil fungus Rhizoctonia solani anastomosis group 3 strain Rhs1AP. Genome Announc. 2014, 2, e01072-14. [Google Scholar] [CrossRef]
- Zheng, A.; Lin, R.; Zhang, D.; Qin, P.; Xu, L.; Ai, P.; Ding, L.; Wang, Y.; Chen, Y.; Liu, Y. The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat. Commun. 2013, 4, 1424. [Google Scholar] [CrossRef]
- Taheri, P.; Gnanamanickam, S.; Höfte, M. Characterization, genetic structure, and pathogenicity of Rhizoctonia spp. associated with rice sheath diseases in India. Phytopathology 2007, 97, 373–383. [Google Scholar] [CrossRef]
- Ferrucho, R.L.; Cifuentes, J.M.; Ceresini, P.; García-Domínguez, C. Rhizoctonia solani AG-3PT is the major pathogen associated with potato stem canker and black scurf in Colombia. Agron. Colomb. 2012, 30, 204–213. [Google Scholar]
- Hane, J.K.; Anderson, J.P.; Williams, A.H.; Sperschneider, J.; Singh, K.B. Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8. PLoS Genet. 2014, 10, e1004281. [Google Scholar] [CrossRef]
- Anderson, J.P.; Sperschneider, J.; Win, J.; Kidd, B.; Yoshida, K.; Hane, J.; Saunders, D.G.; Singh, K.B. Comparative secretome analysis of Rhizoctonia solani isolates with different host ranges reveals unique secretomes and cell death inducing effectors. Sci. Rep. 2017, 7, 10410. [Google Scholar] [CrossRef]
- Adhipathi, P.; Singh, V.; Meena, S.C. Virulence diversity of Rhizoctonia solani causing sheath blight disease in rice and its host pathogen interaction. Bioscan 2013, 8, 949–952. [Google Scholar]
- Sandoval, R.F.C.; Cumagun, C.J.R. Phenotypic and molecular analyses of Rhizoctonia spp. associated with rice and other hosts. Microorganisms 2019, 7, 88. [Google Scholar] [CrossRef]
- Wibberg, D.; Andersson, L.; Tzelepis, G.; Rupp, O.; Blom, J.; Jelonek, L.; Pühler, A.; Fogelqvist, J.; Varrelmann, M.; Schlüter, A. Genome analysis of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB revealed high numbers in secreted proteins and cell wall degrading enzymes. BMC Genom. 2016, 17, 245. [Google Scholar] [CrossRef]
- Thon, M.R.; Pan, H.; Diener, S.; Papalas, J.; Taro, A.; Mitchell, T.K.; Dean, R.A. The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol. 2006, 7, R16. [Google Scholar] [CrossRef]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.-R.; Pan, H. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980. [Google Scholar] [CrossRef]
- Dong, S.; Raffaele, S.; Kamoun, S. The two-speed genomes of filamentous pathogens: Waltz with plants. Curr. Opin. Genet. Dev. 2015, 35, 57–65. [Google Scholar] [CrossRef]
- Ma, L.-J.; Fedorova, N.D. A practical guide to fungal genome projects: Strategy, technology, cost and completion. Mycology 2010, 1, 9–24. [Google Scholar] [CrossRef]
- Dhillon, B.; Gill, N.; Hamelin, R.C.; Goodwin, S.B. The landscape of transposable elements in the finished genome of the fungal wheat pathogen Mycosphaerella graminicola. BMC Genom. 2014, 15, 1132. [Google Scholar] [CrossRef] [PubMed]
- Mat Razali, N.; Cheah, B.H.; Nadarajah, K. Transposable Elements Adaptive Role in Genome Plasticity, Pathogenicity and Evolution in Fungal Phytopathogens. Int. J. Mol. Sci. 2019, 20, 3597. [Google Scholar] [CrossRef]
- Nadarajah, K.; Razali, N.M.; Cheah, B.H.; Sahruna, N.S.; Ismail, I.; Tathode, M.; Bankar, K. Draft genome sequence of Rhizoctonia solani anastomosis group 1 subgroup 1A strain 1802/KB isolated from rice. Genome Announc. 2017, 5, e01188-17. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Muszewska, A.; Hoffman-Sommer, M.; Grynberg, M. LTR retrotransposons in fungi. PLoS ONE 2011, 6, e29425. [Google Scholar] [CrossRef]
- Castanera, R.; Lopez-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Perez, G.; Pisabarro, A.G.; Grigoriev, I.V. Transposable elements versus the fungal genome: Impact on whole-genome architecture and transcriptional profiles. PLoS Genet. 2016, 12, e1006108. [Google Scholar] [CrossRef]
- Beauregard, A.; Curcio, M.J.; Belfort, M. The take and give between retrotransposable elements and their hosts. Annu. Rev. Genet. 2008, 42, 587–617. [Google Scholar] [CrossRef]
- Kazazian, H.H. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- van Regenmortel, M.H.; Mahy, B.W. Desk Encyclopedia of Plant and Fungal Virology; Academic Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Amyotte, S.G.; Tan, X.; Pennerman, K.; del Mar Jimenez-Gasco, M.; Klosterman, S.J.; Ma, L.-J.; Dobinson, K.F.; Veronese, P. Transposable elements in phytopathogenic Verticillium spp.: Insights into genome evolution and inter-and intra-specific diversification. BMC Genom. 2012, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Daboussi, M.-J.; Capy, P. Transposable elements in filamentous fungi. Annu. Rev. Microbiol. 2003, 57, 275–299. [Google Scholar] [CrossRef]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef]
- Vukich, M.; Giordani, T.; Natali, L.; Cavallini, A. Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 2009, 9, 150. [Google Scholar] [CrossRef]
- Havecker, E.R.; Gao, X.; Voytas, D.F. The diversity of LTR retrotransposons. Genome Biol. 2004, 5, 225. [Google Scholar] [CrossRef]
- da Silva, L.L.; Moreno, H.L.A.; Correia, H.L.N.; Santana, M.F.; de Queiroz, M.V. Colletotrichum: Species complexes, lifestyle, and peculiarities of some sources of genetic variability. Appl. Microbiol. Biotechnol. 2020, 104, 1891–1904. [Google Scholar] [CrossRef]
- Ma, J.; Devos, K.M.; Bennetzen, J.L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14, 860–869. [Google Scholar] [CrossRef]
- Rep, M. Small proteins of plant-pathogenic fungi secreted during host colonization. FEMS Microbiol. Lett. 2005, 253, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Pederson, S.M.; Kortschak, R.D.; Adelson, D.L. Transposable elements and gene expression during the evolution of amniotes. Mobile DNA 2018, 9, 17. [Google Scholar] [CrossRef]
- Wei, M.; Wang, A.; Liu, Y.; Ma, L.; Niu, X.; Zheng, A. Identification of the novel effector RsIA_NP8 in Rhizoctonia solani AG1 IA that induces cell death and triggers defense responses in non-host plants. Front. Microbiol. 2020, 11, 1115. [Google Scholar] [CrossRef]
- Li, S.; Peng, X.; Wang, Y.; Hua, K.; Xing, F.; Zheng, Y.; Liu, W.; Sun, W.; Wei, S. The effector AGLIP1 in Rhizoctonia solani AG1 IA triggers cell death in plants and promotes disease development through inhibiting PAMP-triggered immunity in Arabidopsis thaliana. Front. Microbiol. 2019, 10, 2228. [Google Scholar] [CrossRef]
- Dölfors, F.; Holmquist, L.; Dixelius, C.; Tzelepis, G. A LysM effector protein from the basidiomycete Rhizoctonia solani contributes to virulence through suppression of chitin-triggered immunity. Mol. Genet. Genom. 2019, 294, 1211–1218. [Google Scholar] [CrossRef]
- Abdul Malik, N.A.; Kumar, I.S.; Nadarajah, K. Elicitor and receptor molecules: Orchestrators of plant defense and immunity. Int. J. Mol. Sci. 2020, 21, 963. [Google Scholar] [CrossRef] [PubMed]
- Depotter, J.R.; Zuo, W.; Hansen, M.; Zhang, B.; Xu, M.; Doehlemann, G. Effectors with different gears: Divergence of Ustilago maydis effector genes is associated with their temporal expression pattern during plant infection. J. Fungi 2021, 7, 16. [Google Scholar]
- King, B.C.; Waxman, K.D.; Nenni, N.V.; Walker, L.P.; Bergstrom, G.C.; Gibson, D.M. Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol. Biofuels 2011, 4, 4. [Google Scholar] [CrossRef]
- Couturier, M.; Navarro, D.; Olivé, C.; Chevret, D.; Haon, M.; Favel, A.; Lesage-Meessen, L.; Henrissat, B.; Coutinho, P.M.; Berrin, J.-G. Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genom. 2012, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2008, 37, D233–D238. [Google Scholar] [CrossRef]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef]
- Ospina-Giraldo, M.D.; Griffith, J.G.; Laird, E.W.; Mingora, C. The CAZyome of Phytophthora spp.: A comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genom. 2010, 11, 525. [Google Scholar] [CrossRef]
- Garron, M.-L.; Cygler, M. Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 2010, 20, 1547–1573. [Google Scholar] [CrossRef]
- Sharma, A.; Tewari, R.; Rana, S.S.; Soni, R.; Soni, S.K. Cellulases: Classification, methods of determination and industrial applications. Appl. Biochem. Biotechnol. 2016, 179, 1346–1380. [Google Scholar] [CrossRef]
- Yennamalli, R.M.; Rader, A.J.; Kenny, A.J.; Wolt, J.D.; Sen, T.Z. Endoglucanases: Insights into thermostability for biofuel applications. Biotechnol. Biofuels 2013, 6, 136. [Google Scholar] [CrossRef]
- Yamamoto, N.; Wang, Y.; Lin, R.; Liang, Y.; Liu, Y.; Zhu, J.; Wang, L.; Wang, S.; Liu, H.; Deng, Q. Integrative transcriptome analysis discloses the molecular basis of a heterogeneous fungal phytopathogen complex, Rhizoctonia solani AG-1 subgroups. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.; De Lorono, I.; Foster, S.; Li, D.; Johnstone, K.; Ashby, A. Evidence for a role of cutinase in pathogenicity of Pyrenopeziza brassicae on brassicas. Physiol. Mol. Plant Pathol. 2000, 57, 63–75. [Google Scholar] [CrossRef]
- Ramzi, A.B.; Me, M.L.C.; Ruslan, U.S.; Baharum, S.N.; Muhammad, N.A.N. Insight into plant cell wall degradation and pathogenesis of Ganoderma boninense via comparative genome analysis. PeerJ 2019, 7, e8065. [Google Scholar] [CrossRef]
- Farrer, R.A.; Fisher, M.C. Describing genomic and epigenomic traits underpinning emerging fungal pathogens. Adv. Genet. 2017, 100, 73–140. [Google Scholar] [PubMed]
- Sang, H.; Hulvey, J.P.; Green, R.; Xu, H.; Im, J.; Chang, T.; Jung, G. A Xenobiotic Detoxification Pathway through Transcriptional Regulation in Filamentous Fungi. mBio 2018, 9, e00457-18. [Google Scholar] [CrossRef]
- Patel, D.D.; Patel, A.K.; Parmar, N.R.; Shah, T.M.; Patel, J.B.; Pandya, P.R.; Joshi, C.G. Microbial and Carbohydrate Active Enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet. Gene 2014, 545, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Jeong, Y.-U.; Kong, W.-S. Genome Sequencing and Carbohydrate-Active Enzyme (CAZyme) Repertoire of the White Rot Fungus Flammulina elastica. Int. J. Mol. Sci. 2018, 19, 2379. [Google Scholar] [CrossRef] [PubMed]
- Brenelli, L.B.; Persinoti, G.F.; Cairo, J.P.L.F.; Liberato, M.V.; Gonçalves, T.A.; Otero, I.V.; Mainardi, P.H.; Felby, C.; Sette, L.D.; Squina, F.M. Novel redox-active enzymes for ligninolytic applications revealed from multiomics analyses of Peniophora sp. CBMAI 1063, a laccase hyper-producer strain. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Hu, J.; Arantes, V.; Pribowo, A.; Gourlay, K.; Saddler, J.N. Substrate factors that influence the synergistic interaction of AA9 and cellulases during the enzymatic hydrolysis of biomass. Energy Environ. Sci. 2014, 7, 2308–2315. [Google Scholar] [CrossRef]
- DeYoung, B.J.; Innes, R.W. Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 2006, 7, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Armenta, S.; Moreno-Mendieta, S.; Sánchez-Cuapio, Z.; Sánchez, S.; Rodríguez-Sanoja, R. Advances in molecular engineering of carbohydrate-binding modules. Proteins 2017, 85, 1602–1617. [Google Scholar] [CrossRef]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Tang, H. PacBio sequencing reveals transposable elements as a key contributor to genomic plasticity and virulence variation in Magnaporthe oryzae. Mol. Plant 2017, 10, 1465–1468. [Google Scholar] [CrossRef]
- Omrane, S.; Audéon, C.; Ignace, A.; Duplaix, C.; Aouini, L.; Kema, G.; Walker, A.-S.; Fillinger, S. Plasticity of the MFS1 promoter leads to multidrug resistance in the wheat pathogen Zymoseptoria tritici. MSphere 2017, 2. [Google Scholar] [CrossRef]
- de Jonge, R.; Bolton, M.D.; Kombrink, A.; van den Berg, G.C.; Yadeta, K.A.; Thomma, B.P. Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 2013, 23, 1271–1282. [Google Scholar] [CrossRef]
- Kirkland, T.N.; Muszewska, A.; Stajich, J.E. Analysis of transposable elements in Coccidioides species. J. Fungi 2018, 4, 13. [Google Scholar] [CrossRef]
- Chadha, S.; Sharma, M. Transposable elements as stress adaptive capacitors induce genomic instability in fungal pathogen Magnaporthe oryzae. PLoS ONE 2014, 9, e94415. [Google Scholar] [CrossRef]
- Barber, E.A.; Liu, Z.; Smith, S.R. Organic Contaminant Biodegradation by Oxidoreductase Enzymes in Wastewater Treatment. Microorganisms 2020, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H. Discovery and characterization of new O-methyltransferase from the genome of the lignin-degrading fungus Phanerochaete chrysosporium for enhanced lignin degradation. Enzym. Microb. Technol. 2016, 82, 66–73. [Google Scholar]
- Ismail, I.A.; Able, A.J. Secretome analysis of virulent Pyrenophora teres f. teres isolates. Proteomics 2016, 16, 2625–2636. [Google Scholar] [CrossRef] [PubMed]
- Shim, W.-B.; Sagaram, U.S.; Choi, Y.-E.; So, J.; Wilkinson, H.H.; Lee, Y.-W. FSR1 is essential for virulence and female fertility in Fusarium verticillioides and F. graminearum. Mol. Plant-Microbe Interact. 2006, 19, 725–733. [Google Scholar] [CrossRef]
- Soanes, D.M.; Alam, I.; Cornell, M.; Wong, H.M.; Hedeler, C.; Paton, N.W.; Rattray, M.; Hubbard, S.J.; Oliver, S.G.; Talbot, N.J. Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. PLoS ONE 2008, 3, e2300. [Google Scholar] [CrossRef]
- Cantu, D.; Vicente, A.R.; Labavitch, J.M.; Bennett, A.B.; Powell, A.L. Strangers in the matrix: Plant cell walls and pathogen susceptibility. Trends Plant Sci. 2008, 13, 610–617. [Google Scholar] [CrossRef]
- Park, J.; Lappe, M.; Teichmann, S.A. Mapping protein family interactions: Intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast. J. Mol. Biol. 2001, 307, 929–938. [Google Scholar] [CrossRef]
- Devi, B.S.R.; Kim, Y.-J.; Sathiyamoorthy, S.; Khorolragchaa, A.; Gayathri, S.; Parvin, S.; Yang, D.-U.; Selvi, S.K.; Lee, O.R.; Lee, S. Classification and characterization of putative cytochrome P450 genes from Panax ginseng CA Meyer. Biochemistry 2011, 76, 1347–1359. [Google Scholar]
- Moktali, V.; Park, J.; Fedorova-Abrams, N.D.; Park, B.; Choi, J.; Lee, Y.-H.; Kang, S. Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genom. 2012, 13, 525. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Hub, A.; Group, W.P.W. AmiGO: Online access to ontology and annotation data. Bioinformatics 2008, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Daboussi, M.J. Fungal transposable elements: Generators of diversity and genetic tools. J. Genet. 1996, 75, 325. [Google Scholar] [CrossRef]
- Črešnar, B.; Petrič, Š. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 2011, 1814, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hunt, M.; Tsai, I.J. Inferring synteny between genome assemblies: A systematic evaluation. BMC Bioinform. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-Y.; Jeon, J.; Kim, K.-T.; Cheong, K.; Song, H.; Choi, G.; Koh, J.; Opiyo, S.; Zuo, S.; Madhav, M.S. Comparative genome analyses of four rice-infecting isolates of Rhizoctonia solani belonging to anastomosis group 1-intraspecific group IA (RsAG1-IA). Plant Health 2019. [Google Scholar]
- Losada, L.; Pakala, S.B.; Fedorova, N.D.; Joardar, V.; Shabalina, S.A.; Hostetler, J.; Pakala, S.M.; Zafar, N.; Thomas, E.; Rodriguez-Carres, M. Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 2014, 352, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; Silva, N.D.; Martinez, M.C.; Helder Pedro, A.D.Y.; Hassani-Pak, K.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2019, 48, D613–D620. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A Web Resource For Automated Carbohydrate-active Enzyme Annotation. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome | Draft AG1-IAT | China AG1-IA | AG1-IB | AG3 | AG8 |
|---|---|---|---|---|---|
| Reference | Nadarajah et al. 2017 | Zheng et al. 2013 | Wibberg et al. 2012 | Cubeta et al. 2014 | Hane et al. 2014 |
| Host range | Isolated from rice | Rice, maize, soybean | Lettuce, bean, cabbage and carrot | Solanacae family | Cereals, brassicas, legumes |
| Sequencing Method | Illumina Mi Seq | Illumina Genome Analyser II | Illumina Mi-Seq | Sanger (4-, 10-, and 40-kb insert sizes), GS-FLX 454 (fragment and 20-kb mate pair) | Illumina |
| Assembly method | Velvet Medusa | SOAP denovo 1.05 | Newbler Assembly version 2.8 | Celera Assembler 5.1, CLC | SOAP denovo 1.05, SSPACE 2.1, GapCloser 1.2 |
| Assembly size (Mbp) | 28.93 | 36.94 | 42.80 | 51.71 | 48.83 |
| Accession number | GCA_001899475.2 | GCA_000334115.1 | LN679100:LN679996 | GCA_000524645.1 | GCA_000695385.1 |
| N50 | 420,418 | 474,500 | 25,869 | ||
| GC content (%) | 46.99 | 47.61 | 48.04 | 48.41 | 48.83 |
| Number of scaffold | 1517 | 2648 | 897 | 326 | 861 |
| Protein coding genes | 10,037 | 10,489 | 12,616 | 12,726 | 13,964 |
| Repeat Elements (%) | 4.86% | 5.27% | - | - | 10.03 |
| Genome | Tools | % of Repeat Elements | Dominant Repeat |
|---|---|---|---|
| Draft AG1-IA | RepeatScout, RepeatModeller, RepeatMasker and RepBase | 4.86 | Long Terminal Retrotransposon (Gypsy) |
| China AG1-IA | PILER, RepeatScout, RepeatMasker and RepBase | 5.27 | Long Terminal Retrotransposon (Gypsy) |
| AG8 | RepeatScout, CAP3, RepeatMasker and RepBase | 10.03 | Long Terminal Retrotransposon (Gypsy) |
| Anastomosis Group (AG) | Total CAZy | Total no. of Protein-Coding Genes | Percentage of CAZy Out of Protein Coding Genes (%) |
|---|---|---|---|
| Draft AG1-IA | 315 | 10,037 | 3.138388 |
| China AG1-IA | 122 | 10,489 | 1.163123 |
| AG1-IB | 385 | 12,616 | 3.05168 |
| AG3 | 409 | 12,726 | 3.213893 |
| AG8 | 279 | 13,952 | 1.999713 |
| AGs | Draft AG1-IA | China AG1-IA | AG1-IB | AG3 | AG8 |
|---|---|---|---|---|---|
| Glycoside Hydrolase (GH) | GH16 (15) | GH13 (6) | GH28 (25) | GH28 (21) | GH5 (17) |
| GH5 (14) | GH16 (5) | GH16 (20) | GH 16 (16) | GH16 (13) | |
| GH13 (11) | GH5 (15) | GH5 (16) | GH28 (9) | ||
| GH28 (10) | |||||
| Glycosyl Transferases (GT) | GT2 (12) | GT2 (8) | GT2 (12) | GT2 (11) | GT2 (9) |
| GT4 (6) | GT4 (4) | GT4 (7) | GT4 (7) | GT4 (8) | |
| GT1 (6) | GT39 (3) | GT1 (6) | GT1 (5) | GT1 (4) | |
| Polysaccharide Lyases (PL) | PL1 (15) | PL1 (5) | PL1 (23) | PL1 (25) | PL1 (14) |
| PL3 (7) | PL4 (2) | PL3 (19) | PL3 (15) | PL3 (13) | |
| PL4 (7) | PL4 (9) | PL4 (10) | PL4 (3) | ||
| Auxilliary Activities (AA) | AA9 (11) | AA1 (5) | AA9 (11) | AA9 (13) | AA9 (11) |
| AA1 (8) | AA5 (4) | AA1 (10) | AA5 (11) | AA5 (10) | |
| AA5 (6) | AA3 (3) | AA5 (9) | AA1 (11) | AA1 (8) | |
| Carbohydrate Esterases (CE) | CE8 (8) | CE8 (5) | CE5 (9) | CE8 (11) | CE4 (7) |
| CE5 (6) | CE12 (1) | CE8 (8) | CE4 (9) | CE5 (6) | |
| CE4 (5) | CE4 (1) | CE4 (7) | CE5 (6) | CE8 (6) | |
| Carbohdrate-Binding- Molecules (CBM) | CBM13 (6) | CBM13 (3) | CBM1 (8) | CBM1 (9) | CBM1 (10) |
| CBM1 (5) | CBM13 (5) | CBM13 (6) | CBM13 (4) | ||
| CBM48 (1) | CBM50 (1) | CBM5 (1) | CBM50 (1) |
| Anastomosis Group (AG) | Host Range | Category | TE Abundance (%) | Assembly Size (Mbp) |
|---|---|---|---|---|
| Draft AG1-IA | Isolated from rice | Monocot | 2.22 | 28.93 |
| China AG1-IA | Rice, corn, sorghum, bean, soybean, turf grass, camphor, seedlings | Monocot-specific (Narrow) | 3.67 | 36.94 |
| AG1-IB | Rice, bean, soybean, leguminous woody plants, lettuce, hortensia, cabbage | Both monocot & dicot (Intermediate/Broad) | 3.18 | 42.80 |
| AG3 | Solanaceae: Potatoes, tobacco, tomato, egg plant | Dicot-specific (Narrow/Intermediate) | 1.02 | 51.71 |
| AG8 | Poaceae: Cereals, brassicas, legumes | Both monocot & dicot (Broad) | 2.80 | 48.83 |
| Total Number of Proteins | 59,588 |
|---|---|
| Number of Core Othologs | 3936 |
| Number of Single Copy Orthologs | 2758 |
| Cellular Component | Biological Processes | Molecular Function |
|---|---|---|
| Integral component of membrane | Translation | ATP binding |
| Nucleus | Transcription, DNA-templated | Metal ion binding |
| Cytoplasm | Transmembrane transport | DNA binding |
| Intracellular | Metabolic process | Zinc ion binding |
| Ribosome | Carbohydrate metabolic process | Hydrolase |
| Total Number of LCBs | 17,610 |
| Number of LCBs ≥ 1 kbp (mean) | 9697 |
| Number of LCBs ≥ 100 kbp (mean) | 46 |
| Number of Core LCBs | 2272 |
| Sum of mean length of Core LCBs (bp) | 27, 246, 585.6 |
| Number of Core LCBs ≥ 1 kbp (mean) | 2121 |
| Number of Core LCBs ≥ 10 kbp (mean) | 698 |
| Number of Core LCBs ≥ 100 kbp (mean) | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mat Razali, N.; Hisham, S.N.; Kumar, I.S.; Shukla, R.N.; Lee, M.; Abu Bakar, M.F.; Nadarajah, K. Comparative Genomics: Insights on the Pathogenicity and Lifestyle of Rhizoctonia solani. Int. J. Mol. Sci. 2021, 22, 2183. https://doi.org/10.3390/ijms22042183
Mat Razali N, Hisham SN, Kumar IS, Shukla RN, Lee M, Abu Bakar MF, Nadarajah K. Comparative Genomics: Insights on the Pathogenicity and Lifestyle of Rhizoctonia solani. International Journal of Molecular Sciences. 2021; 22(4):2183. https://doi.org/10.3390/ijms22042183
Chicago/Turabian StyleMat Razali, Nurhani, Siti Norvahida Hisham, Ilakiya Sharanee Kumar, Rohit Nandan Shukla, Melvin Lee, Mohd Faizal Abu Bakar, and Kalaivani Nadarajah. 2021. "Comparative Genomics: Insights on the Pathogenicity and Lifestyle of Rhizoctonia solani" International Journal of Molecular Sciences 22, no. 4: 2183. https://doi.org/10.3390/ijms22042183
APA StyleMat Razali, N., Hisham, S. N., Kumar, I. S., Shukla, R. N., Lee, M., Abu Bakar, M. F., & Nadarajah, K. (2021). Comparative Genomics: Insights on the Pathogenicity and Lifestyle of Rhizoctonia solani. International Journal of Molecular Sciences, 22(4), 2183. https://doi.org/10.3390/ijms22042183