
Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke

 ,
, 
 , , , and
, , , and 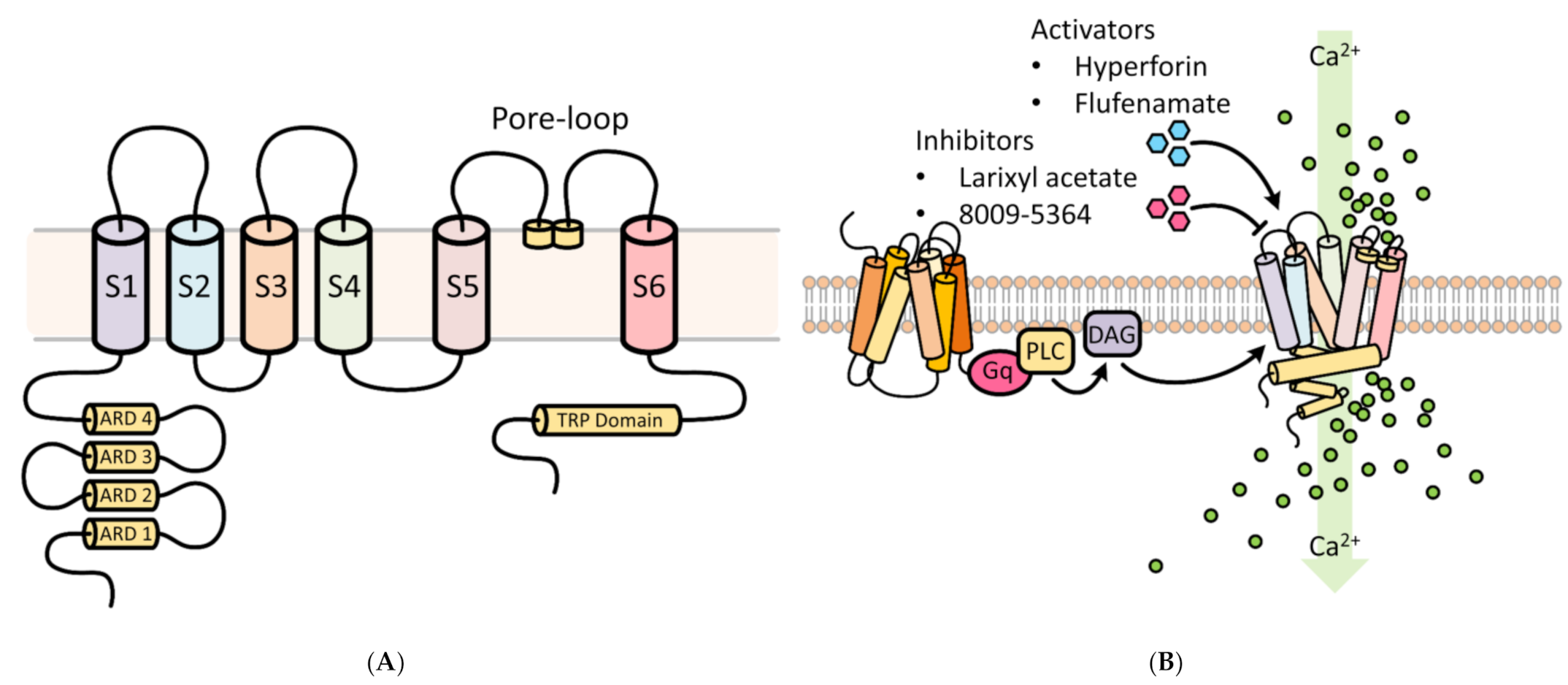{kind=link}
{kind=link}
Abstract
1. Introduction
2. TRPC6
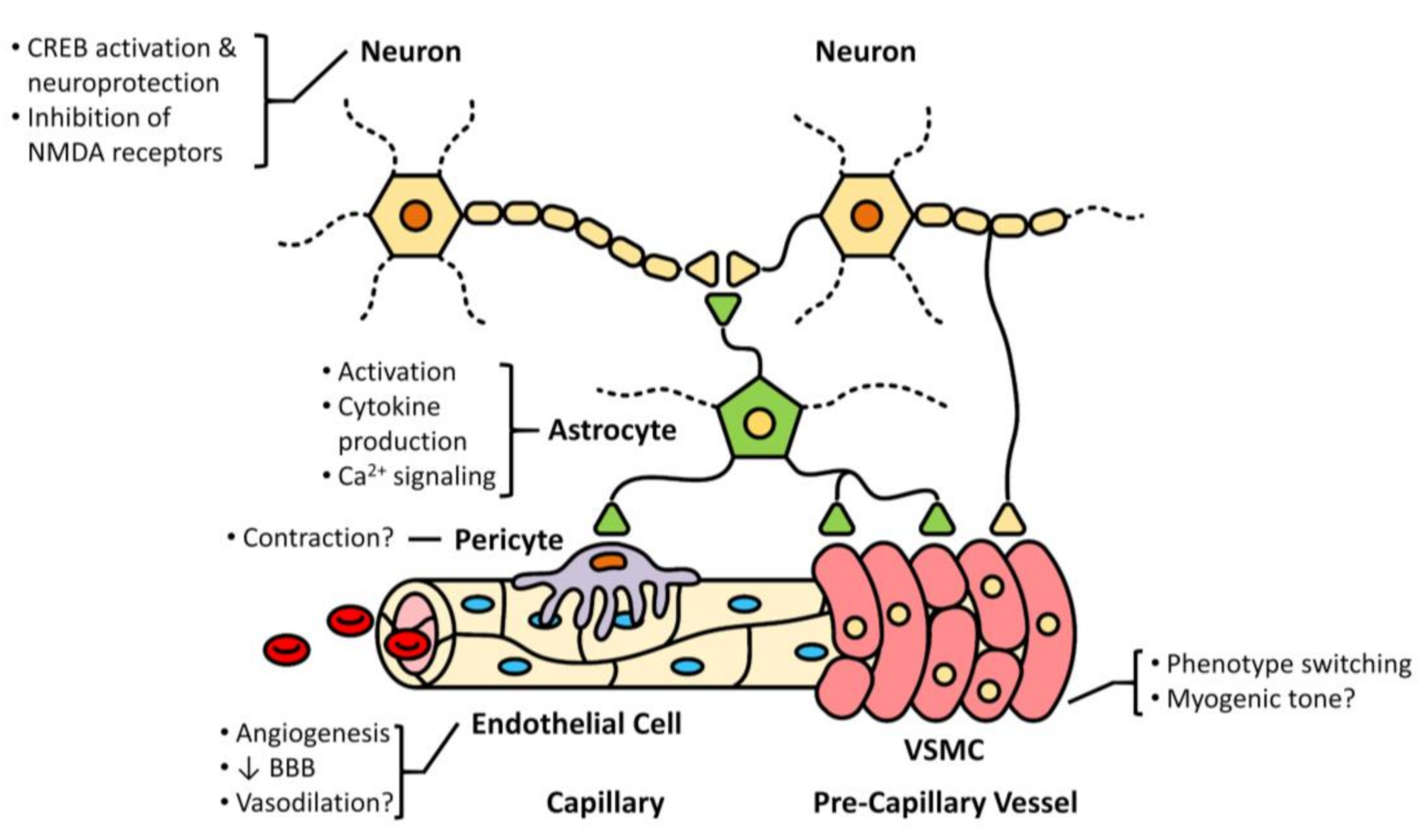
3. Cell-Specific Roles of TRPC6
3.1. Astrocytes
3.2. Neurons
3.3. Endothelial Cells
3.4. Vascular Smooth Muscle Cells (VSMCs)
4. Neurovascular Coupling in Ischemic Stroke
5. Role of Neuronal TRPC 6 in Ischemic Stroke
6. Therapeutic Opportunities
7. Conclusions and Clinical Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Hall, M.J.; Levant, S.; DeFrances, C.J. Hospitalization for Stroke in U.S. Hospitals, 1989–2009; NCHS Data Brief; US Department of Health and Human Services: Washington, DC, USA, 2012; Volume 85, pp. 1–8. [Google Scholar]
- Gorelick, P.B. The global burden of stroke: Persistent and disabling. Lancet Neurol. 2019, 18, 417–418. [Google Scholar] [CrossRef]
- Fifi, J.T.; Mocco, J. COVID-19 related stroke in young individuals. Lancet Neurol. 2020, 19, 713–715. [Google Scholar] [CrossRef]
- Jakobsson, J.; Malm, C.; Furberg, M.; Ekelund, U.; Svensson, M. Physical Activity during the Coronavirus (COVID-19) Pandemic: Prevention of a Decline in Metabolic and Immunological Functions. Front. Sports Act. Living 2020, 2, 57. [Google Scholar] [CrossRef]
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of ischemic and hemorrhagic stroke: Incidence, prevalence, mortality, and risk factors. Neurol. Clin. 2008, 26, 871–895. [Google Scholar] [CrossRef]
- Kim, S.M.; Jung, J.M.; Kim, B.J.; Lee, J.S.; Kwon, S.U. Cilostazol Mono and Combination Treatments in Ischemic Stroke: An Updated Systematic Review and Meta-Analysis. Stroke 2019, 50, 3503–3511. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.K.; Olsen, T.S.; Dehlendorff, C.; Kammersgaard, L.P. Hemorrhagic and ischemic strokes compared: Stroke severity, mortality, and risk factors. Stroke 2009, 40, 2068–2072. [Google Scholar] [CrossRef]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38 (Suppl. 2), 674–676. [Google Scholar] [CrossRef]
- Smith, W.S. Pathophysiology of focal cerebral ischemia: A therapeutic perspective. J. Vasc. Interv. Radiol. 2004, 15, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Liu, R.; Travis, O.K.; Roman, R.J.; Fan, F. Cerebral Autoregulation in Hypertension and Ischemic Stroke: A Mini Review. J. Pharm. Sci. Exp. Pharmacol. 2017, 2017, 21–27. [Google Scholar]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Muralikrishna Adibhatla, R.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hawkins, K.E.; Dore, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 129, 1021–1029. [Google Scholar] [CrossRef]
- Iadecola, C. Regulation of the cerebral microcirculation during neural activity: Is nitric oxide the missing link? Trends Neurosci. 1993, 16, 206–214. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, X.; Lin, X.; Zhang, J.; You, X.; Shao, A. The Role of Transient Receptor Potential Channels in Blood-Brain Barrier Dysfunction after Ischemic Stroke. Biomed. Pharmacother. 2020, 131, 110647. [Google Scholar] [CrossRef]
- Li, H.; Huang, J.; Du, W.; Jia, C.; Yao, H.; Wang, Y. TRPC6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity. J. Neurochem. 2012, 123, 1010–1018. [Google Scholar] [CrossRef]
- Huang, J. TRPC Channels and Stroke. Adv. Exp. Med. Biol. 2017, 976, 61–71. [Google Scholar]
- Du, W.; Huang, J.; Yao, H.; Zhou, K.; Duan, B.; Wang, Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J. Clin. Investig. 2010, 120, 3480–3492. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Z. Higher Peripheral Blood MiR-488 Level Predicts Poor Prognosis of Acute Ischemic Stroke. Clin. Lab. 2020, 66. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhan, X.; He, M.; Wang, J.; Qiu, X. miR-135b levels in the peripheral blood serve as a marker associated with acute ischemic stroke. Exp. Ther. Med. 2020, 19, 3551–3558. [Google Scholar] [CrossRef] [PubMed]
- Montell, C.; Birnbaumer, L.; Flockerzi, V.; Bindels, R.J.; Bruford, E.A.; Caterina, M.J.; Clapham, D.E.; Harteneck, C.; Heller, S.; Julius, D.; et al. A unified nomenclature for the superfamily of TRP cation channels. Mol. Cell 2002, 9, 229–231. [Google Scholar] [CrossRef]
- Dietrich, A.; Chubanov, V.; Kalwa, H.; Rost, B.R.; Gudermann, T. Cation channels of the transient receptor potential superfamily: Their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol. Ther. 2006, 112, 744–760. [Google Scholar] [CrossRef]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harbor Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Gualdani, R.; Gailly, P. How TRPC Channels Modulate Hippocampal Function. Int. J. Mol. Sci. 2020, 21, 3915. [Google Scholar] [CrossRef] [PubMed]
- Hirschler-Laszkiewicz, I.; Tong, Q.; Conrad, K.; Zhang, W.; Flint, W.W.; Barber, A.J.; Barber, D.L.; Cheung, J.Y.; Miller, B.A. TRPC3 activation by erythropoietin is modulated by TRPC6. J. Biol. Chem. 2009, 284, 4567–4581. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Kamouchi, M.; Philipp, S.; Flockerzi, V.; Wissenbach, U.; Mamin, A.; Raeymaekers, L.; Eggermont, J.; Droogmans, G.; Nilius, B. Properties of heterologously expressed hTRP3 channels in bovine pulmonary artery endothelial cells. J. Physiol. 1999, 518, 345–358. [Google Scholar] [CrossRef]
- Zitt, C.; Obukhov, A.G.; Strubing, C.; Zobel, A.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J. Cell Biol. 1997, 138, 1333–1341. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.J.; Qiu, J.; Ronnekleiv, O.K. TRPCing around the hypothalamus. Front. Neuroendocrinol. 2018, 51, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [PubMed]
- Putney, J.W.; Tomita, T. Phospholipase C signaling and calcium influx. Adv. Biol. Regul. 2012, 52, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Chai, Z.; Chen, Y.; Wang, C. beta-arrestin-1: Bridging GPCRs to active TRP channels. Channels (Austin) 2017, 11, 357–359. [Google Scholar] [CrossRef]
- Liu, C.H.; Gong, Z.; Liang, Z.L.; Liu, Z.X.; Yang, F.; Sun, Y.J.; Ma, M.L.; Wang, Y.J.; Ji, C.R.; Wang, Y.H.; et al. Arrestin-biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat. Commun. 2017, 8, 14335. [Google Scholar] [CrossRef]
- Patel, A.; Sharif-Naeini, R.; Folgering, J.R.; Bichet, D.; Duprat, F.; Honore, E. Canonical TRP channels and mechanotransduction: From physiology to disease states. Pflugers Arch. Eur. J. Physiol. 2010, 460, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Dyachenko, V.; Husse, B.; Rueckschloss, U.; Isenberg, G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 2009, 45, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Lu, S.; Wang, Y.; Kim, T.; Mehta, D.; Wang, Y. The role of mechanical tension on lipid raft dependent PDGF-induced TRPC6 activation. Biomaterials 2014, 35, 2868–2877. [Google Scholar] [CrossRef]
- Nikolaev, Y.A.; Cox, C.D.; Ridone, P.; Rohde, P.R.; Cordero-Morales, J.F.; Vasquez, V.; Laver, D.R.; Martinac, B. Mammalian TRP ion channels are insensitive to membrane stretch. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef]
- Shirakawa, H.; Katsumoto, R.; Iida, S.; Miyake, T.; Higuchi, T.; Nagashima, T.; Nagayasu, K.; Nakagawa, T.; Kaneko, S. Sphingosine-1-phosphate induces Ca2+ signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 2017, 65, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Wang, Y.; Li, X.; Wu, L.; Wang, Y. TRPC6 expression in neurons is differentially regulated by NR2A- and NR2B-containing NMDA receptors. J. Neurochem. 2017, 143, 282–293. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Chen, M.; Lu, T.J.; Chen, X.J.; Zhou, Y.; Chen, Q.; Feng, X.Y.; Xu, L.; Duan, W.H.; Xiong, Z.Q. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 2008, 39, 3042–3048. [Google Scholar] [CrossRef]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef]
- Beskina, O.; Miller, A.; Mazzocco-Spezzia, A.; Pulina, M.V.; Golovina, V.A. Mechanisms of interleukin-1beta-induced Ca2+ signals in mouse cortical astrocytes: Roles of store- and receptor-operated Ca2+ entry. Am. J. Physiol. Cell Physiol. 2007, 293, C1103–C1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, Z.; Liu, Q.; Sun, W.; Jiang, B.; Yang, K.; Li, J.; Gong, Y.; Liu, Q.; Liu, D.; et al. Nongenetic optical modulation of neural stem cell proliferation and neuronal/glial differentiation. Biomaterials 2019, 225, 119539. [Google Scholar] [CrossRef]
- Choi, H.J.; Sun, D.; Jakobs, T.C. Astrocytes in the optic nerve head express putative mechanosensitive channels. Mol. Vis. 2015, 21, 749–766. [Google Scholar]
- Fan, Q.; Huang, W.B.; Zhang, X.L. TRPC6: An underlying target for human glaucoma. Int. J. Ophthalmol. 2012, 5, 523–526. [Google Scholar]
- Belkacemi, T.; Niermann, A.; Hofmann, L.; Wissenbach, U.; Birnbaumer, L.; Leidinger, P.; Backes, C.; Meese, E.; Keller, A.; Bai, X.; et al. TRPC1- and TRPC3-dependent Ca2+ signaling in mouse cortical astrocytes affects injury-evoked astrogliosis in vivo. Glia 2017, 65, 1535–1549. [Google Scholar] [CrossRef]
- Zeng, C.; Tian, F.; Xiao, B. TRPC Channels: Prominent Candidates of Underlying Mechanism in Neuropsychiatric Diseases. Mol. Neurobiol. 2016, 53, 631–647. [Google Scholar] [CrossRef]
- Zeitler, S.; Schumacher, F.; Monti, J.; Anni, D.; Guhathakurta, D.; Kleuser, B.; Friedland, K.; Fejtova, A.; Kornhuber, J.; Rhein, C. Acid Sphingomyelinase Impacts Canonical Transient Receptor Potential Channels 6 (TRPC6) Activity in Primary Neuronal Systems. Cells 2020, 9, 2502. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.; Ma, S.; Mu, F.; Deng, J.; Duan, J.; Xiong, L.; Yin, Y.; Wang, Y.; Xi, M.; et al. The Role of TRPC6 in the Neuroprotection of Calycosin against Cerebral Ischemic Injury. Sci. Rep. 2017, 7, 3039. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, L.; Liu, S.; Wang, X. Tetramethylpyrazine Protects Neurons from Oxygen-Glucose Deprivation-Induced Death. Med. Sci. Monit. 2017, 23, 5277–5282. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Hatcher, J.T.; Chen, Q.H.; Chen, L.; Wurster, R.D.; Chan, S.L.; Cheng, Z. Deletion of TRPC6 Attenuates NMDA Receptor-Mediated Ca2+ Entry and Ca2+-Induced Neurotoxicity Following Cerebral Ischemia and Oxygen-Glucose Deprivation. Front. Neurosci. 2017, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Gao, P.; Cui, Y.; Li, Q.; Li, Y.; Lu, Z.; Ma, H.; Zhao, Y.; Li, L.; Sun, F.; et al. Low-glucose-sensitive TRPC6 dysfunction drives hypoglycemia-induced cognitive impairment in diabetes. Clin. Transl. Med. 2020, 10, e205. [Google Scholar] [CrossRef]
- Tai, Y.; Feng, S.; Du, W.; Wang, Y. Functional roles of TRPC channels in the developing brain. Pflug. Arch. Eur. J. Physiol. 2009, 458, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.B.; Stein, V.; Bonhoeffer, T.; Lohmann, C. Endogenous brain-derived neurotrophic factor triggers fast calcium transients at synapses in developing dendrites. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sukumaran, P.; Bandyopadhyay, B.C.; Singh, B.B. Physiological Function and Characterization of TRPCs in Neurons. Cells 2014, 3, 455–475. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Pu, K.; Duan, W.; Chen, H.; Chen, L.; Wang, Y. Involvement of Akt/CREB signaling pathways in the protective effect of EPA against interleukin-1beta-induced cytotoxicity and BDNF down-regulation in cultured rat hippocampal neurons. BMC Neurosci. 2018, 19, 52. [Google Scholar] [CrossRef]
- Jia, Y.; Zhou, J.; Tai, Y.; Wang, Y. TRPC channels promote cerebellar granule neuron survival. Nat. Neurosci. 2007, 10, 559–567. [Google Scholar] [CrossRef]
- Brown, R.C.; Wu, L.; Hicks, K.; O’Neil, R.G. Regulation of blood-brain barrier permeability by transient receptor potential type C and type v calcium-permeable channels. Microcirculation 2008, 15, 359–371. [Google Scholar] [CrossRef]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Singh, I.; Knezevic, N.; Ahmmed, G.U.; Kini, V.; Malik, A.B.; Mehta, D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J. Biol. Chem. 2007, 282, 7833–7843. [Google Scholar] [CrossRef]
- Samapati, R.; Yang, Y.; Yin, J.; Stoerger, C.; Arenz, C.; Dietrich, A.; Gudermann, T.; Adam, D.; Wu, S.; Freichel, M.; et al. Lung endothelial Ca2+ and permeability response to platelet-activating factor is mediated by acid sphingomyelinase and transient receptor potential classical 6. Am. J. Respir. Crit. Care Med. 2012, 185, 160–170. [Google Scholar] [CrossRef]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; James, A.F.; Foster, R.R.; Hancox, J.C.; Bates, D.O. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.; Hildebrandt, E.; Ryan, M.J.; Granger, J.P.; Drummond, H.A. Pressure-induced constriction of the middle cerebral artery is abolished in TrpC6 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H42–H50. [Google Scholar] [CrossRef]
- Wang, Q.; Leo, M.D.; Narayanan, D.; Kuruvilla, K.P.; Jaggar, J.H. Local coupling of TRPC6 to ANO1/TMEM16A channels in smooth muscle cells amplifies vasoconstriction in cerebral arteries. Am. J. Physiol. Cell Physiol. 2016, 310, C1001–C1009. [Google Scholar] [CrossRef]
- Inoue, R.; Jensen, L.J.; Jian, Z.; Shi, J.; Hai, L.; Lurie, A.I.; Henriksen, F.H.; Salomonsson, M.; Morita, H.; Kawarabayashi, Y.; et al. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ. Res. 2009, 104, 1399–1409. [Google Scholar] [CrossRef]
- Salinet, A.S.; Silva, N.C.; Caldas, J.; de Azevedo, D.S.; de-Lima-Oliveira, M.; Nogueira, R.C.; Conforto, A.B.; Texeira, M.J.; Robinson, T.G.; Panerai, R.B.; et al. Impaired cerebral autoregulation and neurovascular coupling in middle cerebral artery stroke: Influence of severity? J. Cereb. Blood Flow. Metab. 2019, 39, 2277–2285. [Google Scholar] [CrossRef]
- Lin, W.H.; Hao, Q.; Rosengarten, B.; Leung, W.H.; Wong, K.S. Impaired neurovascular coupling in ischaemic stroke patients with large or small vessel disease. Eur. J. Neurol. 2011, 18, 731–736. [Google Scholar] [CrossRef]
- Girouard, H.; Iadecola, C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. 2006, 100, 328–335. [Google Scholar] [CrossRef]
- Bundo, M.; Inao, S.; Nakamura, A.; Kato, T.; Ito, K.; Tadokoro, M.; Kabeya, R.; Sugimoto, T.; Kajita, Y.; Yoshida, J. Changes of Neural Activity Correlate with the Severity of Cortical Ischemia in Patients with Unilateral Major Cerebral Artery Occlusion. Stroke 2002, 33, 61–66. [Google Scholar] [CrossRef]
- McConnell, H.L.; Li, Z.; Woltjer, R.L.; Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem. Int. 2019, 128, 70–84. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Raman-Nair, J.; Lacoste, B. Structural and Functional Remodeling of the Brain Vasculature Following Stroke. Front. Physiol. 2020, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral Vascular Disease and Neurovascular Injury in Ischemic Stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Tanaka, T.; Mangmool, S.; Nishiyama, K.; Nishimura, A. Canonical Transient Receptor Potential Channels and Vascular Smooth Muscle Cell Plasticity. J. Lipid Atheroscler 2020, 9, 124–139. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Shimauchi, T.; Oda, S.; Tanaka, T.; Nishiyama, K.; Nishimura, A.; Birnbaumer, L.; Mori, Y.; Nishida, M. TRPC6 regulates phenotypic switching of vascular smooth muscle cells through plasma membrane potential-dependent coupling with PTEN. FASEB J. 2019, 33, 9785–9796. [Google Scholar] [CrossRef]
- Hariharan, A.; Weir, N.; Robertson, C.; He, L.; Betsholtz, C.; Longden, T.A. The Ion Channel and GPCR Toolkit of Brain Capillary Pericytes. Front. Cell Neurosci. 2020, 14, 601324. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Underly, R.G.; Levy, M.; Hartmann, D.A.; Grant, R.I.; Watson, A.N.; Shih, A.Y. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J. Neurosci. 2017, 37, 129. [Google Scholar] [CrossRef]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Nuñez, V.; Kaplan, L.; Granger, A.J.; Bistrong, K.; Zucker, H.L.; Kumar, P.; Sabatini, B.L.; Gu, C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 2020, 579, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A. Upstream current for a downstream flow. Nat. Neurosci. 2017, 20, 631–633. [Google Scholar] [CrossRef]
- Xu, P.; Xu, J.; Li, Z.; Yang, Z. Expression of TRPC6 in renal cortex and hippocampus of mouse during postnatal development. PLoS ONE 2012, 7, e38503. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, J.C.; Fu, J.; Chen, F.; Wang, J.; Wu, Z.L.; Yuan, S.Y. Hyperforin attenuates brain damage induced by transient middle cerebral artery occlusion (MCAO) in rats via inhibition of TRPC6 channels degradation. J. Cereb. Blood Flow. Metab. 2013, 33, 253–262. [Google Scholar] [CrossRef]
- Shukla, A.; Bosenberg, M.W.; MacPherson, M.B.; Butnor, K.J.; Heintz, N.H.; Pass, H.I.; Carbone, M.; Testa, J.R.; Mossman, B.T. Activated cAMP response element binding protein is overexpressed in human mesotheliomas and inhibits apoptosis. Am. J. Pathol. 2009, 175, 2197–2206. [Google Scholar] [CrossRef]
- Wang, J.; Sun, R.; Li, Z.; Pan, Y. Combined bone marrow stromal cells and oxiracetam treatments ameliorates acute cerebral ischemia/reperfusion injury through TRPC6. Acta Biochim. Biophys. Sin. (Shanghai) 2019, 51, 767–777. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, X.; Zhou, T.; Cheng, X.; Lin, Y. IL-17A contributes to brain ischemia reperfusion injury through calpain-TRPC6 pathway in mice. Neuroscience 2014, 274, 419–428. [Google Scholar] [CrossRef]
- Zhang, E.; Liao, P. Brain transient receptor potential channels and stroke. J. Neurosci. Res. 2015, 93, 1165–1183. [Google Scholar] [CrossRef]
- Chen, X.; Lu, M.; He, X.; Ma, L.; Birnbaumer, L.; Liao, Y. TRPC3/6/7 Knockdown Protects the Brain from Cerebral Ischemia Injury via Astrocyte Apoptosis Inhibition and Effects on NF-кB Translocation. Mol. Neurobiol. 2017, 54, 7555–7566. [Google Scholar] [CrossRef]
- Hoffmann, U.; Sheng, H.; Ayata, C.; Warner, D.S. Anesthesia in Experimental Stroke Research. Transl. Stroke Res. 2016, 7, 358–367. [Google Scholar] [CrossRef]
- Higashi, Y.; Aratake, T.; Shimizu, S.; Shimizu, T.; Saito, M. [Brain zinc dyshomeostasis and glial cells in ischemic stroke]. Nihon Yakurigaku Zasshi Folia Pharmacol. Jpn. 2019, 154, 138–142. [Google Scholar] [CrossRef]
- Wang, Z.; do Carmo, J.M.; da Silva, A.A.; Fu, Y.; Hall, J.E. Mechanisms of Synergistic Interactions of Diabetes and Hypertension in Chronic Kidney Disease: Role of Mitochondrial Dysfunction and ER Stress. Curr. Hypertens. Rep. 2020, 22, 15. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, J.P.; Göritz, C.; Tatarishvili, J.; Dias, D.O.; Smith, E.M.; Lindvall, O.; Kokaia, Z.; Frisén, J. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science 2014, 346, 237–241. [Google Scholar] [CrossRef]
- Prikhodko, V.; Chernyuk, D.; Sysoev, Y.; Zernov, N.; Okovityi, S.; Popugaeva, E. Potential Drug Candidates to Treat TRPC6 Channel Deficiencies in the Pathophysiology of Alzheimer’s Disease and Brain Ischemia. Cells 2020, 9, 2531. [Google Scholar] [CrossRef]
- Leuner, K.; Kazanski, V.; Muller, M.; Essin, K.; Henke, B.; Gollasch, M.; Harteneck, C.; Muller, W.E. Hyperforin--a key constituent of St. John’s wort specifically activates TRPC6 channels. FASEB J. 2007, 21, 4101–4111. [Google Scholar] [CrossRef]
- Sell, T.S.; Belkacemi, T.; Flockerzi, V.; Beck, A. Protonophore properties of hyperforin are essential for its pharmacological activity. Sci. Rep. 2014, 4, 7500. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, F.; Zhang, J.; Wang, T.; Wei, X.; Wu, J.; Feng, Y.; Dai, Z.; Wu, Q. Neuroprotective effect of resveratrol on ischemia/reperfusion injury in rats through TRPC6/CREB pathways. J. Mol. Neurosci. 2013, 50, 504–513. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, J.; Chen, F.; Lin, Y. Neuroprotectin D1 attenuates brain damage induced by transient middle cerebral artery occlusion in rats through TRPC6/CREB pathways. Mol. Med. Rep. 2013, 8, 543–550. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, J.; Liu, G.; Chen, F.; Lin, Y. Neuroprotection by (-)-epigallocatechin-3-gallate in a rat model of stroke is mediated through inhibition of endoplasmic reticulum stress. Mol. Med. Rep. 2014, 9, 69–76. [Google Scholar] [CrossRef]
- Sossin, W.S.; Barker, P.A. Something old, something new: BDNF-induced neuron survival requires TRPC channel function. Nat. Neurosci. 2007, 10, 537–538. [Google Scholar] [CrossRef]
- Liu, L.; Gu, L.; Chen, M.; Zheng, Y.; Xiong, X.; Zhu, S. Novel Targets for Stroke Therapy: Special Focus on TRPC Channels and TRPC6. Front. Aging Neurosci. 2020, 12, 70. [Google Scholar] [CrossRef]
- Chrostek, M.R.; Fellows, E.G.; Crane, A.T.; Grande, A.W.; Low, W.C. Efficacy of stem cell-based therapies for stroke. Brain Res. 2019, 1722, 146362. [Google Scholar] [CrossRef]
- Li, W.; Yang, F.; Gao, J.; Tang, Y.; Wang, J.; Pan, Y. Over-Expression of TRPC6 via CRISPR Based Synergistic Activation Mediator in BMSCs Ameliorates Brain Injury in a Rat Model of Cerebral Ischemia/Reperfusion. Neuroscience 2019, 415, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Montecinos-Oliva, C.; Schuller, A.; Parodi, J.; Melo, F.; Inestrosa, N.C. Effects of tetrahydrohyperforin in mouse hippocampal slices: Neuroprotection, long-term potentiation and TRPC channels. Curr. Med. Chem. 2014, 21, 3494–3506. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Hafner, S.; Urban, N.; Schaefer, M. Discovery and characterization of a positive allosteric modulator of transient receptor potential canonical 6 (TRPC6) channels. Cell Calcium 2019, 78, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Vemana, H.P.; Karim, Z.A.; Conlon, C.; Khasawneh, F.T. A critical role for the transient receptor potential channel type 6 in human platelet activation. PLoS ONE 2015, 10, e0125764. [Google Scholar] [CrossRef]
- Paez Espinosa, E.V.; Lin, O.A.; Karim, Z.A.; Alshbool, F.Z.; Khasawneh, F.T. Mouse transient receptor potential channel type 6 selectively regulates agonist-induced platelet function. Biochem. Biophys. Rep. 2019, 20, 100685. [Google Scholar] [CrossRef]
- Hassock, S.R.; Zhu, M.X.; Trost, C.; Flockerzi, V.; Authi, K.S. Expression and role of TRPC proteins in human platelets: Evidence that TRPC6 forms the store-independent calcium entry channel. Blood 2002, 100, 2801–2811. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, G.; Gupta, S.; Thielmann, I.; Pleines, I.; Varga-Szabo, D.; May, F.; Mannhalter, C.; Dietrich, A.; Nieswandt, B.; Braun, A. Defective diacylglycerol-induced Ca2+ entry but normal agonist-induced activation responses in TRPC6-deficient mouse platelets. J. Thromb. Haemost. 2012, 10, 419–429. [Google Scholar] [CrossRef]
- Fernandez, M.; Lao-Peregrin, C.; Martin, E.D. Flufenamic acid suppresses epileptiform activity in hippocampus by reducing excitatory synaptic transmission and neuronal excitability. Epilepsia 2010, 51, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lozinskaya, I.; Costell, M.; Lin, Z.; Ball, J.A.; Bernard, R.; Behm, D.J.; Marino, J.P.; Schnackenberg, C.G. Characterization of Small Molecule TRPC3 and TRPC6 agonist and Antagonists. Biophys. J. 2013, 104, 454a. [Google Scholar] [CrossRef]
- Tiapko, O.; Shrestha, N.; Lindinger, S.; Guedes de la Cruz, G.; Graziani, A.; Klec, C.; Butorac, C.; Graier, W.F.; Kubista, H.; Freichel, M.; et al. Lipid-independent control of endothelial and neuronal TRPC3 channels by light. Chem. Sci. 2019, 10, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, S.; Hatano, M.; Takada, Y.; Hino, K.; Kawamura, T.; Tanikawa, J.; Nakagawa, H.; Hase, H.; Nakao, A.; Hirano, M.; et al. Screening of Transient Receptor Potential Canonical Channel Activators Identifies Novel Neurotrophic Piperazine Compounds. Mol. Pharmacol. 2016, 89, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.; Wang, L.; Kwiek, S.; Rademann, J.; Kuebler, W.M.; Schaefer, M. Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Mol. Pharmacol. 2016, 89, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.W.; Sachs, F.; Bush, E.D.; Suchyna, T.M. GsMTx4-D provides protection to the D2.mdx mouse. Neuromuscul. Disord. 2018, 28, 868–877. [Google Scholar] [CrossRef]
- Shaik, J.S.; Poloyac, S.M.; Kochanek, P.M.; Alexander, H.; Tudorascu, D.L.; Clark, R.S.; Manole, M.D. 20-Hydroxyeicosatetraenoic Acid Inhibition by HET0016 Offers Neuroprotection, Decreases Edema, and Increases Cortical Cerebral Blood Flow in a Pediatric Asphyxial Cardiac Arrest Model in Rats. J. Cereb. Blood Flow. Metab. 2015, 35, 1757–1763. [Google Scholar] [CrossRef]
- Poloyac, S.M.; Zhang, Y.; Bies, R.R.; Kochanek, P.M.; Graham, S.H. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J. Cereb. Blood Flow. Metab. 2006, 26, 1551–1561. [Google Scholar] [CrossRef]
- Urban, N.; Hill, K.; Wang, L.; Kuebler, W.M.; Schaefer, M. Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell Calcium 2012, 51, 194–206. [Google Scholar] [CrossRef]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shekhar, S.; Liu, Y.; Wang, S.; Zhang, H.; Fang, X.; Zhang, J.; Fan, L.; Zheng, B.; Roman, R.J.; Wang, Z.; et al. Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 2074. https://doi.org/10.3390/ijms22042074
Shekhar S, Liu Y, Wang S, Zhang H, Fang X, Zhang J, Fan L, Zheng B, Roman RJ, Wang Z, et al. Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke. International Journal of Molecular Sciences. 2021; 22(4):2074. https://doi.org/10.3390/ijms22042074
Chicago/Turabian StyleShekhar, Shashank, Yedan Liu, Shaoxun Wang, Huawei Zhang, Xing Fang, Jin Zhang, Letao Fan, Baoying Zheng, Richard J. Roman, Zhen Wang, and et al. 2021. "Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke" International Journal of Molecular Sciences 22, no. 4: 2074. https://doi.org/10.3390/ijms22042074
APA StyleShekhar, S., Liu, Y., Wang, S., Zhang, H., Fang, X., Zhang, J., Fan, L., Zheng, B., Roman, R. J., Wang, Z., Fan, F., & Booz, G. W. (2021). Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke. International Journal of Molecular Sciences, 22(4), 2074. https://doi.org/10.3390/ijms22042074