
Computational Selectivity Assessment of Protease Inhibitors against SARS-CoV-2

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Sequence and Active Site Comparison
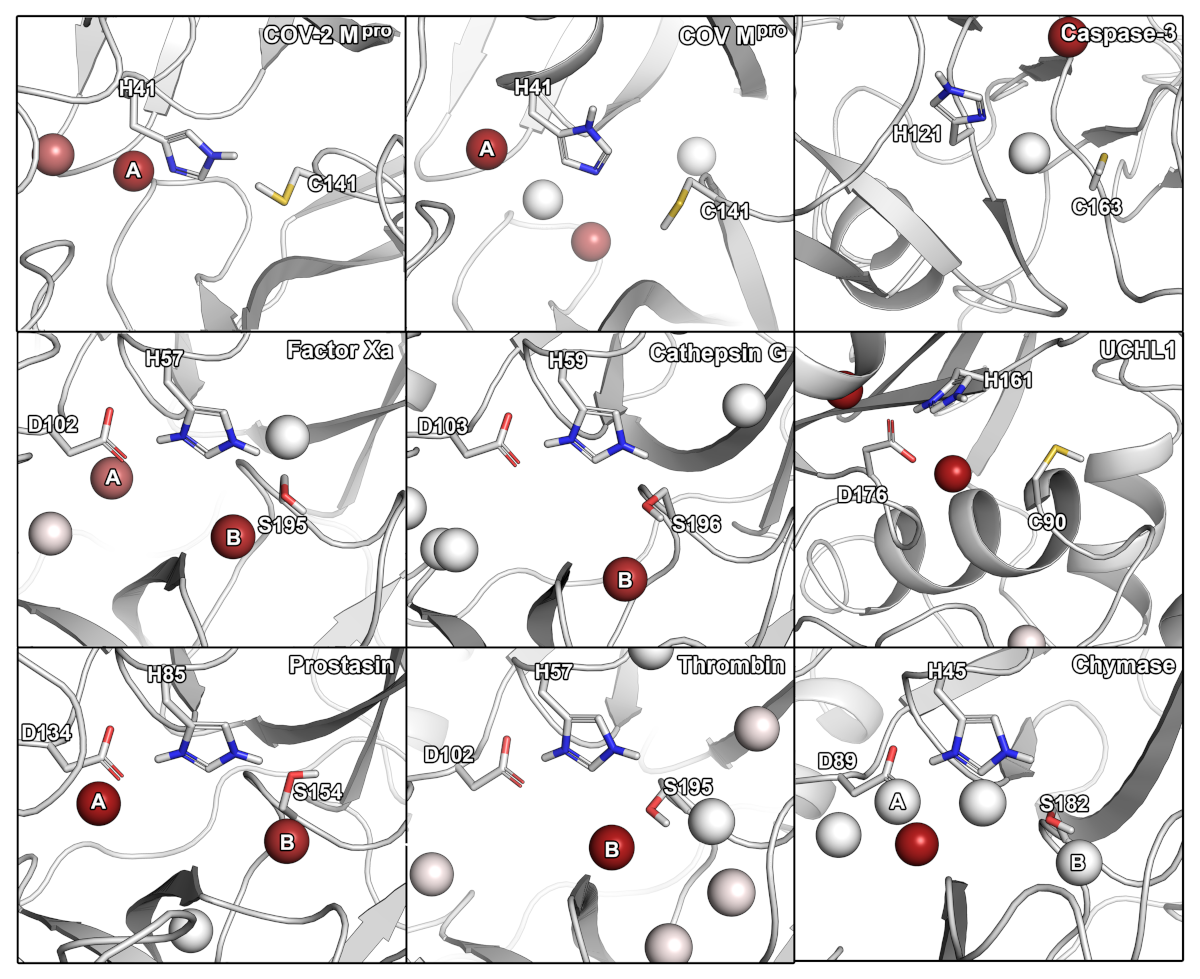
2.2. Hydration and Small-Molecule Hot-Spots
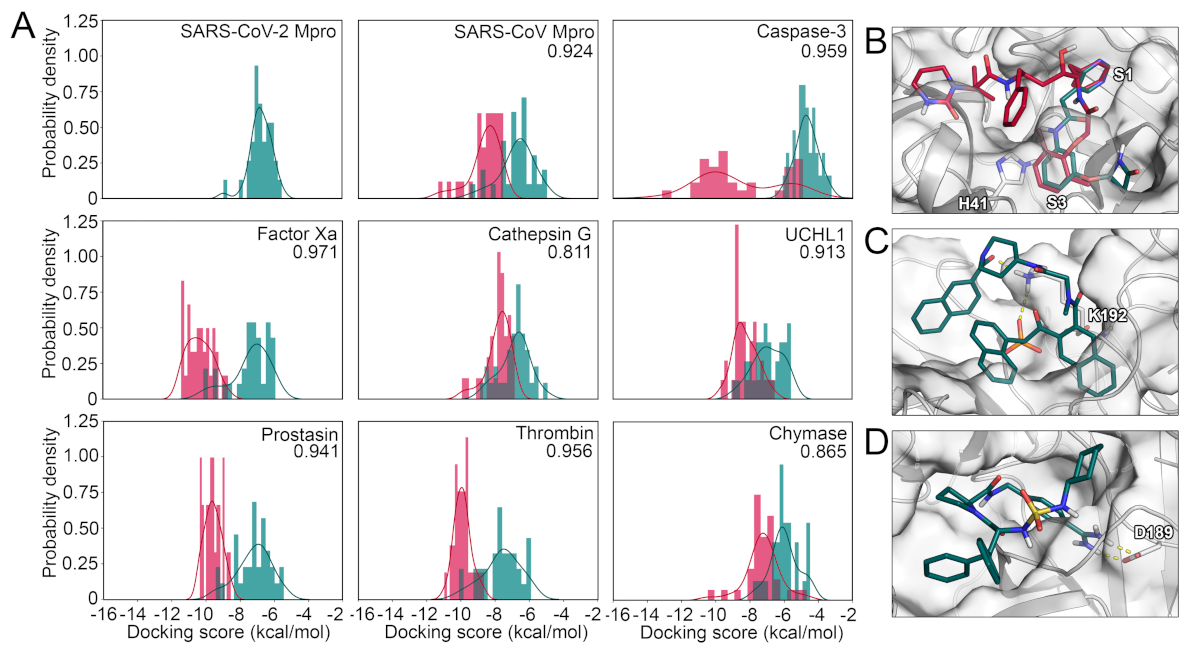
2.3. Protease Selectivity Assessed by Molecular Docking
2.4. Toxicity Profiling
2.5. Selectivity from Different Perspectives
3. Materials and Methods
3.1. Selection of Proteases
3.2. Similarity of Proteins and Active Sites
3.3. Cosolvent MD Simulations
3.4. Classical MD Simulations
3.5. Water Molecules Tracking, Hot-Spots Identification
3.6. Molecular Docking and Validation
3.7. MD and MM/GBSA Post-Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Novel Coronavirus (2019-nCoV) Situation Reports; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Kalil, A.C. Treating COVID-19—Off-Label Drug Use, Compassionate Use, and Randomized Clinical Trials During Pandemics. JAMA 2020, 323, 1897–1898. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- He, J.; Hu, L.; Huang, X.; Wang, C.; Zhang, Z.; Wang, Y.; Zhang, D.; Ye, W. Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs: Insights from structures of protease and inhibitors. Int. J. Antimicrob. Agents 2020, 56, 106055. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef] [PubMed]
- Van Vleet, T.R.; Liguori, M.J.; Lynch, J.J.; Rao, M.; Warder, S. Screening Strategies and Methods for Better Off-Target Liability Prediction and Identification of Small-Molecule Pharmaceuticals. SLAS Discov. 2019, 24, 1–24. [Google Scholar] [CrossRef]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A.; Dobler, M.; Hu, Z.; Smieško, M. OpenVirtualToxLab-A platform for generating and exchanging in silico toxicity data. Toxicol. Lett. 2015, 232, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 1–17. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Smieško, M. Allosteric binding sites on nuclear receptors: Focus on drug efficacy and selectivity. Int. J. Mol. Sci. 2020, 21, 534. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; Carlson, H.A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics. J. Med. Chem. 2016, 59, 10383–10399. [Google Scholar] [CrossRef]
- Kimura, S.R.; Hu, H.P.; Ruvinsky, A.M.; Sherman, W.; Favia, A.D. Deciphering Cryptic Binding Sites on Proteins by Mixed-Solvent Molecular Dynamics. J. Chem. Inf. Model. 2017, 57, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Keretsu, S.; Bhujbal, S.P.; Joo Cho, S. Computational study of paroxetine-like inhibitors reveals new molecular insight to inhibit GRK2 with selectivity over ROCK1. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–376. [Google Scholar] [CrossRef]
- Chaudhury, S.; Gray, J.J. Identification of Structural Mechanisms of HIV-1 Protease Specificity Using Computational Peptide Docking: Implications for Drug Resistance. Structure 2009, 17, 1636–1648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boyd, S.E.; Garcia De La Banda, M.; Pike, R.N.; Whisstock, J.C.; Rudy, G.B. PoPS: A computational tool for modeling and predicting protease specificity. Proc. IEEE Comp. Syst. Bioinform. Conf. 2004, 372–381. [Google Scholar] [CrossRef]
- Weill, N.; Rognan, D. Alignment-free ultra-high-throughput comparison of druggable protein-ligand binding sites. J. Chem. Inf. Model. 2010, 50, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, N.; Gabdoulline, R.R.; Nilges, M.; Wade, R.C. Classification of protein sequences by homology modeling and quantitative analysis of electrostatic similarity. Protein. Struct. Funct. Genet. 1999, 37, 379–387. [Google Scholar] [CrossRef]
- Perona, J.J.; Craik, C.S. Structural basis of substrate specificity in the serine proteases. Protein. Sci. 1995, 4, 337–360. [Google Scholar] [CrossRef]
- Tan, Y.S.; Spring, D.R.; Abell, C.; Verma, C.S. The Application of Ligand-Mapping Molecular Dynamics Simulations to the Rational Design of Peptidic Modulators of Protein-Protein Interactions. J. Chem. Ther. Comput. 2015, 11, 3199–3210. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, B.; Lill, M.A. WATsite2.0 with PyMOL Plugin: Hydration Site Prediction and Visualization BT—Protein Function Prediction: Methods and Protocols; Springer: New York, NY, USA, 2017; pp. 123–134. [Google Scholar] [CrossRef]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and evolutionary analysis indicate that the sars-COV-2 mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.R.; Gomez-Puertas, P.; Leo-Macias, A.; Lopez-Romero, P.; Lopez-Viñas, E.; Morreale, A.; Murcia, M.; Wang, K. Computational approaches to model ligand selectivity in drug design. Curr. Top. Med. Chem. 2006, 6, 41–55. [Google Scholar] [CrossRef]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. A cross docking pipeline for improving pose prediction and virtual screening performance. J. Comput.-Aided Mol. Des. 2018, 32, 163–173. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Anson, B.D.; Weaver, J.G.R.; Ackerman, M.J.; Akinsete, O.; Henry, K.; January, C.T.; Badley, A.D. Blockade of HERG channels by HIV protease inhibitors. Lancet (Lond. UK) 2005, 365, 682–686. [Google Scholar] [CrossRef]
- Chen, Y.; Shoichet, B.K. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat. Chem. Biol. 2009, 5, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.; Wang, H.; Luan, B. In Silico Exploration of the Molecular Mechanism of Clinically Oriented Drugs for Possibly Inhibiting SARS-CoV-2’s Main Protease. J. Phys. Chem. Lett. 2020, 11, 4413–4420. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsuyama, S.; Hoshino, T.; Yamamoto, N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, X.; Wang, X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020, 47, 119–121. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, R.; Cheke, R.; Ambhore, J.; Shinde, S. The Molecular Docking Study of Potential Drug Candidates Showing Anti-COVID-19 Activity by Exploring of Therapeutic Targets of SARS-CoV-2. Eurasian J. Med. Oncol. 2020, 4, 185–195. [Google Scholar] [CrossRef]
- Vatansever, E.; Yang, K.; Kratch, K.; Drelich, A.; Cho, C.; Mellott, D.; Xu, S.; Tseng, C.; Liu, W. Bepridil is potent against SARS-CoV-2 In Vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Eleftheriou, P.; Amanatidou, D.; Petrou, A.; Geronikaki, A. In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules 2020, 25, 2529. [Google Scholar] [CrossRef] [PubMed]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Consortium T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed]
- Luchko, T.; Gusarov, S.; Roe, D.R.; Simmerling, C.; Case, D.A.; Tuszynski, J.; Kovalenko, A. Three-Dimensional Molecular Theory of Solvation Coupled with Molecular Dynamics in Amber. J. Chem. Ther. Comput. 2010, 6, 607–624. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution-Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Walker, R.C.; Cheatham, T.E.; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Darden, T. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Comput. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Magdziarz, T.; Mitusińska, K.; Bzówka, M.; Raczyńska, A.; Stańczak, A.; Banas, M.; Bagrowska, W.; Góra, A. AQUA-DUCT 1.0: Structural and functional analysis of macromolecules from an intramolecular voids perspective. Bioinformatics 2020, 36, 2599–2601. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Empereur-Mot, C.; Zagury, J.F.; Montes, M. Screening Explorer—An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, SC, Austin TX, USA, 15 November 2015; pp. 41–53. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger, LCC. Maestro Small-Molecule Drug Discovery Suite 2019-3; Schrödinger, LCC: New York, NY, USA, 2019. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Anti-Target a | Consequence of Inhibition |
|---|---|---|---|
| SARS-CoV-2 Mpro | Viral replication | - | antiviral activity |
| SARS-CoV Mpro | Viral replication | no | antiviral activity |
| Caspase-3 | Apoptosis | yes | interference with development |
| Factor Xa | Coagulation | no | prevention of coagulopathies |
| Cathepsin G | Immune system | yes | interference with immune response |
| UCHL1 | Protein degradation | yes | interference with development and homeostasis |
| Prostasin | Sodium balance | yes | alters homeostasis |
| Thrombin | Coagulation | no | prevention of coagulopathies |
| Chymase | Vasoconstriction | yes | interference with blood pressure |
| Protein | Catalytic | Global | AS RMSD | Site | Fold | Similarity |
|---|---|---|---|---|---|---|
| Residues | Identity a | (Å)b | Similarity c | Index d | ||
| SARS-CoV Mpro | H41, C145 | 96.1% | 0.2 | 0.91 | / | 0.90 |
| Caspase-3 | H121, C163 | 11.6% | 1.9 | 0.67 | / | −0.74 |
| Factor Xa | H57, D102, S195 | 11.6 % | 1.8 | 0.71 | all- | −0.67 |
| Cathepsin G | H59, D103, S196 | 14.5% | 1.8 | 0.61 | all- | −0.71 |
| UCHL1 | C90, H161, D176 | 15.7% | 2.8 | 0.74 | / | −0.98 |
| Prostasin | H85, D134, S154 | 13.1% | 1.6 | 0.69 | all- | 0.26 |
| Thrombin | H57, D102, S195 | 12.5% | 1.8 | 0.74 | all- | 0.31 |
| Chymase | H45, D89, S182 | 19.0% | 1.9 | 0.64 | all- | −0.49 |
| Protein | Docking | Cosolvents | Hydration | Site Similarity a |
|---|---|---|---|---|
| SARS-CoV Mpro | ** | * | ** | *** |
| Caspase-3 | none | none | none | * |
| Factor Xa | ** | ** | * | * |
| Cathepsin G | *** | * | none | * |
| UCHL1 | *** | * | * | none |
| Prostasin | ** | none | * | ** |
| Thrombin | ** | none | * | ** |
| Chymase | ** | * | * | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, A.; Sellner, M.; Mitusińska, K.; Bzówka, M.; Lill, M.A.; Góra, A.; Smieško, M. Computational Selectivity Assessment of Protease Inhibitors against SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 2065. https://doi.org/10.3390/ijms22042065
Fischer A, Sellner M, Mitusińska K, Bzówka M, Lill MA, Góra A, Smieško M. Computational Selectivity Assessment of Protease Inhibitors against SARS-CoV-2. International Journal of Molecular Sciences. 2021; 22(4):2065. https://doi.org/10.3390/ijms22042065
Chicago/Turabian StyleFischer, André, Manuel Sellner, Karolina Mitusińska, Maria Bzówka, Markus A. Lill, Artur Góra, and Martin Smieško. 2021. "Computational Selectivity Assessment of Protease Inhibitors against SARS-CoV-2" International Journal of Molecular Sciences 22, no. 4: 2065. https://doi.org/10.3390/ijms22042065
APA StyleFischer, A., Sellner, M., Mitusińska, K., Bzówka, M., Lill, M. A., Góra, A., & Smieško, M. (2021). Computational Selectivity Assessment of Protease Inhibitors against SARS-CoV-2. International Journal of Molecular Sciences, 22(4), 2065. https://doi.org/10.3390/ijms22042065