
Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts

 , ,
, ,  and
and 
Abstract
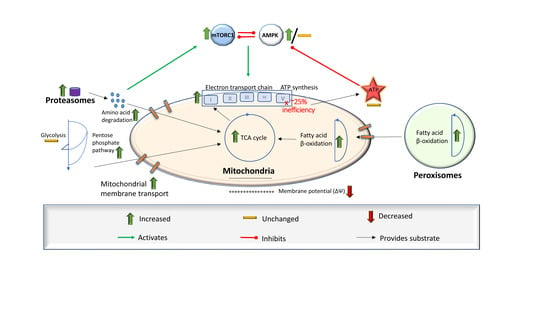
1. Introduction
1.1. Background
1.2. Provisioning the Mitochondria
1.3. Alternative Sources of Oxidisable Substrates Than Carbohydrates
1.4. Investigating Fuel Source Preference in ME/CFS Lymphoblasts
2. Results
2.1. Global Changes in ME/CFS Lymphoblast Transcriptomes and Proteomes
2.1.1. Transcriptomes
2.1.2. Proteomes
2.2. Confirming Previous Protein-Level Expression Results and Examining Transcript Levels in Key Mitochondrial Pathways
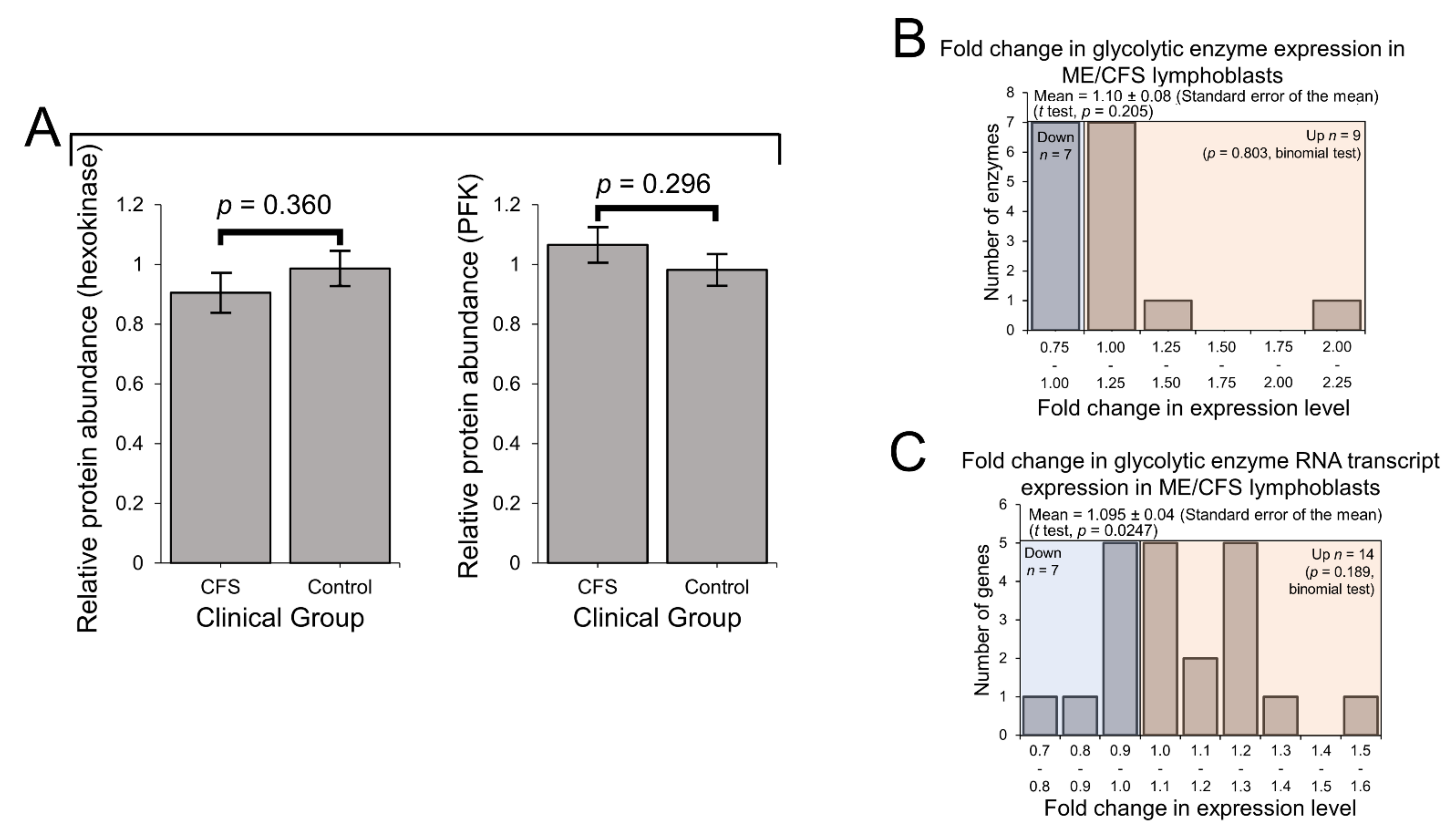
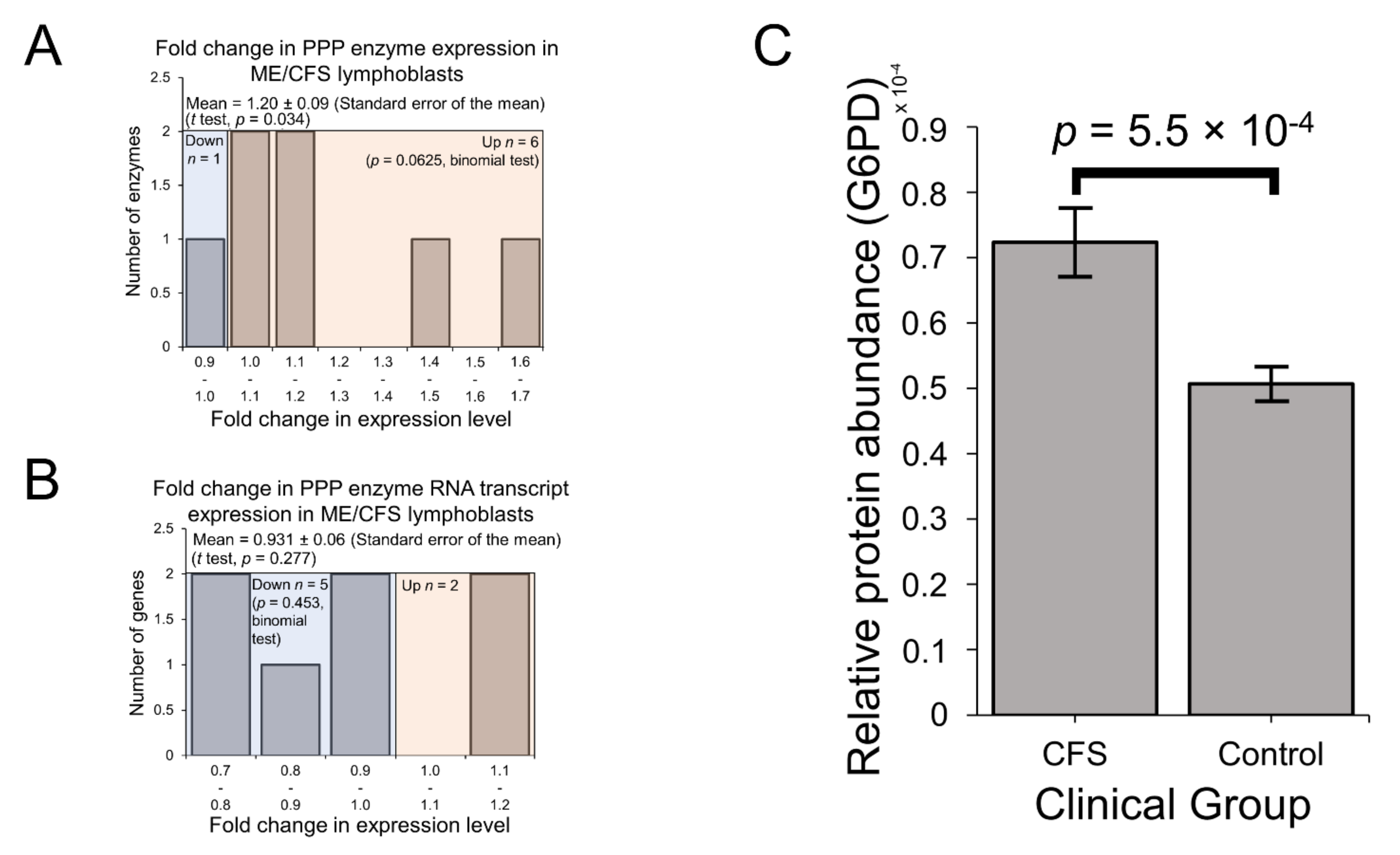
2.3. Expression of Enzymes Involved in Carbohydrate Catabolism by Glycolysis and the Pentose Phosphate Pathway (PPP)
2.4. Enzymes Involved in Mitochondrial and Fatty Acid β-Oxidation Are Elevated in Their Expression
2.5. Expression of Enzymes Involved in the Mitochondrial Utilisation of Glutamine, BCAAs and Essential Amino Acids Is Elevated in ME/CFS Lymphoblasts
2.6. Expression of Proteasome Subunits Is Elevated in ME/CFS Lymphoblasts
3. Discussion
3.1. Preferential Fatty Acid β-Oxidation and Dysregulated Intracellular Energy Stress Signaling
3.2. Dysregulation of Glutamine Metabolism
3.3. Dysregulated Branched-Chain Amino Acid- and Protein-Degradative Pathways
4. Materials and Methods
4.1. Participant Cohort Recruitment, Composition and Subsets
4.1.1. Proteomics
4.1.2. Transcriptomics
4.1.3. AMPK Activity Assay
4.2. Immortalisation, Growth, Storage and Counting of Lymphoblast Cell Lines
4.3. Whole-Cell Proteome Analysis
4.4. RNA Extraction from Lymphoblasts
4.5. RNA Sequencing
4.6. Confirmatory Semi-Quantitative Western Blotting
4.7. Confirmatory qRT-PCR
4.8. AMPK Activity Assay (ACC1/2 Phosphorylation State)
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Falk Hvidberg, M.; Brinth, L.S.; Olesen, A.V.; Petersen, K.D.; Ehlers, L. The Health-Related Quality of Life for Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS ONE 2015, 10, e0132421. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, D.; Annesley, S.J.; Allan, C.Y.; Sanislav, O.; Lidbury, B.A.; Lewis, D.P.; Fisher, P.R. An isolated Complex V inefficiency and dysregulated mitochondrial function in immortalized lymphocytes from ME/CFS patients. Int. J. Mol. Sci. 2020, 21, 1074. [Google Scholar] [CrossRef]
- Armstrong, C.W.; McGregor, N.R.; Sheedy, J.R.; Buttfield, I.; Butt, H.L.; Gooley, P.R. NMR metabolic profiling of serum identifies amino acid disturbances in chronic fatigue syndrome. Clin. Chim. Acta 2012, 413, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.; Butt, H.; Gooley, P.R. Metabolic profiling reveals anomalous energy metabolism and oxidative stress pathways in chronic fatigue syndrome patients. Metabolomics 2015, 11, 1626–1639. [Google Scholar] [CrossRef]
- Yamano, E.; Sugimoto, M.; Hirayama, A.; Kume, S.; Yamato, M.; Jin, G.; Tajima, S.; Goda, N.; Iwai, K.; Fukuda, S.; et al. Index markers of chronic fatigue syndrome with dysfunction of TCA and urea cycles. Sci. Rep. 2016, 6, 34990. [Google Scholar] [CrossRef]
- Fluge, O.; Mella, O.; Bruland, O.; Risa, K.; Dyrstad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Rosland, G.V.; Fossa, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef]
- Mandarano, A.H.; Maya, J.; Giloteaux, L.; Peterson, D.L.; Maynard, M.; Gottschalk, C.G.; Hanson, M.R. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J. Clin. Investig. 2019. [Google Scholar] [CrossRef]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef]
- Tomas, C.; Elson, J.L.; Newton, J.L.; Walker, M. Substrate utilisation of cultured skeletal muscle cells in patients with CFS. Sci. Rep. 2020, 10, 18232. [Google Scholar] [CrossRef]
- Germain, A.; Barupal, D.K.; Levine, S.M.; Hanson, M.R. Comprehensive Circulatory Metabolomics in ME/CFS Reveals Disrupted Metabolism of Acyl Lipids and Steroids. Metabolites 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Horecker, B.L. The pentose phosphate pathway. J. Biol. Chem. 2002, 277, 47965–47971. [Google Scholar] [CrossRef]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol. Biosyst. 2017, 13, 371–379. [Google Scholar] [CrossRef]
- Tapiero, H.; Mathe, G.; Couvreur, P.; Tew, K.D., II. Glutamine and glutamate. Biomed. Pharm. 2002, 56, 446–457. [Google Scholar] [CrossRef]
- Krebs, H.A. Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem. J. 1935, 29, 1951–1969. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef]
- Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Bessa, M.; Plaitakis, A. Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J. Neurosci. Res. 2005, 79, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Abrego, J.; Gunda, V.; Vernucci, E.; Shukla, S.K.; King, R.J.; Dasgupta, A.; Goode, G.; Murthy, D.; Yu, F.; Singh, P.K. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017, 400, 37–46. [Google Scholar] [CrossRef]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Szakal, D.; Barupal, D.K.; Lee, B.; Che, X.; Williams, B.L.; Kahn, E.J.R.; Ukaigwe, J.E.; Bateman, L.; Klimas, N.G.; Komaroff, A.L.; et al. Insights into myalgic encephalomyelitis/chronic fatigue syndrome phenotypes through comprehensive metabolomics. Sci. Rep. 2018, 8, 10056. [Google Scholar] [CrossRef]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Prospective Biomarkers from Plasma Metabolomics of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Implicate Redox Imbalance in Disease Symptomatology. Metabolites 2018, 8. [Google Scholar] [CrossRef]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.P.; Butt, H.L.; Gooley, P.R. The association of fecal microbiota and fecal, blood serum and urine metabolites in myalgic encephalomyelitis/chronic fatigue syndrome. Metabolomics 2017, 13. [Google Scholar] [CrossRef]
- Mensah, F.F.K.; Armstrong, C.W.; Reddy, V.; Bansal, A.S.; Berkovitz, S.; Leandro, M.J.; Cambridge, G. CD24 Expression and B Cell Maturation Shows a Novel Link with Energy Metabolism: Potential Implications for Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Immunol. 2018, 9, 2421. [Google Scholar] [CrossRef]
- Brown, A.E.; Jones, D.E.; Walker, M.; Newton, J.L. Abnormalities of AMPK activation and glucose uptake in cultured skeletal muscle cells from individuals with chronic fatigue syndrome. PLoS ONE 2015, 10, e0122982. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kejariwal, A.; Guo, N.; Mi, H.; Campbell, M.J.; Muruganujan, A.; Lazareva-Ulitsky, B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006, 34, W645–W650. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef]
- Hardcastle, S.L.; Brenu, E.W.; Johnston, S.; Nguyen, T.; Huth, T.; Wong, N.; Ramos, S.; Staines, D.; Marshall-Gradisnik, S. Characterisation of cell functions and receptors in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME). BMC Immunol. 2015, 16, 35. [Google Scholar] [CrossRef]
- Curriu, M.; Carrillo, J.; Massanella, M.; Rigau, J.; Alegre, J.; Puig, J.; Garcia-Quintana, A.M.; Castro-Marrero, J.; Negredo, E.; Clotet, B.; et al. Screening NK-, B- and T-cell phenotype and function in patients suffering from Chronic Fatigue Syndrome. J. Transl. Med. 2013, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Allman, D.; Pillai, S. Peripheral B cell subsets. Curr. Opin. Immunol. 2008, 20, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.R.; Sah, V.P.; Glembotski, C.C.; Brown, J.H. The Rho effector, PKN, regulates ANF gene transcription in cardiomyocytes through a serum response element. Am. J. Physiol Heart Circ. Physiol. 2000, 278, H1769–H1774. [Google Scholar] [CrossRef]
- Israel, B.F.; Gulley, M.; Elmore, S.; Ferrini, S.; Feng, W.H.; Kenney, S.C. Anti-CD70 antibodies: A potential treatment for EBV+ CD70-expressing lymphomas. Mol. Cancer Ther. 2005, 4, 2037–2044. [Google Scholar] [CrossRef]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010, 17, 249–261. [Google Scholar] [CrossRef]
- Ricciardi, S.; Manfrini, N.; Alfieri, R.; Calamita, P.; Crosti, M.C.; Gallo, S.; Muller, R.; Pagani, M.; Abrignani, S.; Biffo, S. The Translational Machinery of Human CD4(+) T Cells Is Poised for Activation and Controls the Switch from Quiescence to Metabolic Remodeling. Cell Metab. 2018, 28, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Weisel, F.J.; Mullett, S.J.; Elsner, R.A.; Menk, A.V.; Trivedi, N.; Luo, W.; Wikenheiser, D.; Hawse, W.F.; Chikina, M.; Smita, S.; et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol. 2020, 21, 331–342. [Google Scholar] [CrossRef]
- Benatti, U.; Morelli, A.; Frascio, M.; Melloni, E.; Salamino, F.; Sparatore, B.; Pontremoli, S.; De Flora, A. Glucose 6-phosphate dehydrogenase activity in membranes of erythrocytes from normal individuals and subjects with Mediterranean G6PD deficiency. Biochem. Biophys Res. Commun. 1978, 85, 1318–1324. [Google Scholar] [CrossRef]
- Xia, C.; Fu, Z.; Battaile, K.P.; Kim, J.P. Crystal structure of human mitochondrial trifunctional protein, a fatty acid beta-oxidation metabolon. Proc. Natl. Acad. Sci. USA 2019, 116, 6069–6074. [Google Scholar] [CrossRef]
- Taylor, W.A.; Mejia, E.M.; Mitchell, R.W.; Choy, P.C.; Sparagna, G.C.; Hatch, G.M. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS ONE 2012, 7, e48628. [Google Scholar] [CrossRef]
- Salazar, D.; Zhang, L.; deGala, G.D.; Frerman, F.E. Expression and characterization of two pathogenic mutations in human electron transfer flavoprotein. J. Biol Chem. 1997, 272, 26425–26433. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef]
- Everett, L.; Galli, A.; Crabb, D. The role of hepatic peroxisome proliferator-activated receptors (PPARs) in health and disease. Liver 2000, 20, 191–199. [Google Scholar] [CrossRef]
- Zeng, J.; Deng, S.; Wang, Y.; Li, P.; Tang, L.; Pang, Y. Specific Inhibition of Acyl-CoA Oxidase-1 by an Acetylenic Acid Improves Hepatic Lipid and Reactive Oxygen Species (ROS) Metabolism in Rats Fed a High Fat Diet. J. Biol. Chem. 2017, 292, 3800–3809. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Moon, Y.A.; Park, S.W.; Cheng, D.; Kwon, H.J.; Horton, J.D. Induced polymerization of mammalian acetyl-CoA carboxylase by MIG12 provides a tertiary level of regulation of fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 9626–9631. [Google Scholar] [CrossRef]
- Jayakumar, A.; Tai, M.H.; Huang, W.Y.; al-Feel, W.; Hsu, M.; Abu-Elheiga, L.; Chirala, S.S.; Wakil, S.J. Human fatty acid synthase: Properties and molecular cloning. Proc. Natl. Acad. Sci. USA 1995, 92, 8695–8699. [Google Scholar] [CrossRef] [PubMed]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Zhang, J.; Frerman, F.E.; Kim, J.J. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. USA 2006, 103, 16212–16217. [Google Scholar] [CrossRef] [PubMed]
- Besrat, A.; Polan, C.E.; Henderson, L.M. Mammalian metabolism of glutaric acid. J. Biol. Chem. 1969, 244, 1461–1467. [Google Scholar] [CrossRef]
- Duran, R.V.; Hall, M.N. Glutaminolysis feeds mTORC1. Cell Cycle 2012, 11, 4107–4108. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell. 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Zhen, H.; Kitaura, Y.; Kadota, Y.; Ishikawa, T.; Kondo, Y.; Xu, M.; Morishita, Y.; Ota, M.; Ito, T.; Shimomura, Y. mTORC1 is involved in the regulation of branched-chain amino acid catabolism in mouse heart. FEBS Open Bio 2016, 6, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Lopez-Maside, L.; Donapetry-Garcia, C.; Fernandez-Fernandez, C.; Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 2017, 49, 1005–1028. [Google Scholar] [CrossRef]
- Zhenyukh, O.; Civantos, E.; Ruiz-Ortega, M.; Sanchez, M.S.; Vazquez, C.; Peiro, C.; Egido, J.; Mas, S. High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic. Biol Med. 2017, 104, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Neishabouri, S.H.; Hutson, S.M.; Davoodi, J. Chronic activation of mTOR complex 1 by branched chain amino acids and organ hypertrophy. Amino Acids 2015, 47, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Sweatt, A.J.; Lanoue, K.F. Branched-chain [corrected] amino acid metabolism: Implications for establishing safe intakes. J. Nutr. 2005, 135, 1557S–1564S. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.K.; Gonzalez, A.; Hall, M.N. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb. Perspect Biol. 2015, 7, a019141. [Google Scholar] [CrossRef]
- Sweetman, E.; Kleffmann, T.; Edgar, C.; de Lange, M.; Vallings, R.; Tate, W. A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J. Transl. Med. 2020, 18, 365. [Google Scholar] [CrossRef]
- Tomas, C.; Brown, A.E.; Newton, J.L.; Elson, J.L. Mitochondrial complex activity in permeabilised cells of chronic fatigue syndrome patients using two cell types. PeerJ 2019, 7, e6500. [Google Scholar] [CrossRef]
- Missailidis, D.; Sanislav, O.; Allan, C.Y.; Annesley, S.J.; Fisher, P.R. Cell-Based Blood Biomarkers for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2020, 21, 1142. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jorgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Peng, T.; Golub, T.R.; Sabatini, D.M. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol. Cell Biol. 2002, 22, 5575–5584. [Google Scholar] [CrossRef]
- Brown, N.F.; Stefanovic-Racic, M.; Sipula, I.J.; Perdomo, G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism 2007, 56, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Dang, C.V. Glutamine Skipping the Q into Mitochondria. Trends Mol. Med. 2020, 26, 6–7. [Google Scholar] [CrossRef]
- Duran, R.V.; MacKenzie, E.D.; Boulahbel, H.; Frezza, C.; Heiserich, L.; Tardito, S.; Bussolati, O.; Rocha, S.; Hall, M.N.; Gottlieb, E. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 2013, 32, 4549–4556. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell. Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- de Vega, W.C.; Vernon, S.D.; McGowan, P.O. DNA methylation modifications associated with chronic fatigue syndrome. PLoS ONE 2014, 9, e104757. [Google Scholar] [CrossRef]
- de Vega, W.C.; Herrera, S.; Vernon, S.D.; McGowan, P.O. Epigenetic modifications and glucocorticoid sensitivity in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). BMC Med. Genom. 2017, 10, 11. [Google Scholar] [CrossRef]
- de Vega, W.C.; Erdman, L.; Vernon, S.D.; Goldenberg, A.; McGowan, P.O. Integration of DNA methylation & health scores identifies subtypes in myalgic encephalomyelitis/chronic fatigue syndrome. Epigenomics 2018, 10, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.S.; Oltra, E.; Sarria, L.; Rose, N.; Beljanski, V.; Fletcher, M.A.; Klimas, N.G.; Nathanson, L. Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-associated DNA methylation patterns. PLoS ONE 2018, 13, e0201066. [Google Scholar] [CrossRef]
- Brenu, E. Methylation Profile of CD4+ T Cells in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Clin. Cell. Immunol. 2014, 05. [Google Scholar] [CrossRef]
- Almenar-Perez, E.; Ovejero, T.; Sanchez-Fito, T.; Espejo, J.A.; Nathanson, L.; Oltra, E. Epigenetic Components of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Uncover Potential Transposable Element Activation. Clin. Ther. 2019, 41, 675–698. [Google Scholar] [CrossRef]
- Helliwell, A.M.; Sweetman, E.C.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Changes in DNA methylation profiles of myalgic encephalomyelitis/chronic fatigue syndrome patients reflect systemic dysfunctions. Clin. Epigenetics 2020, 12, 167. [Google Scholar] [CrossRef]
- Harris, R.A.; Hawes, J.W.; Popov, K.M.; Zhao, Y.; Shimomura, Y.; Sato, J.; Jaskiewicz, J.; Hurley, T.D. Studies on the regulation of the mitochondrial alpha-ketoacid dehydrogenase complexes and their kinases. Adv. Enzym. Regul 1997, 37, 271–293. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Dalle Pezze, P.; Ruf, S.; Sonntag, A.G.; Langelaar-Makkinje, M.; Hall, P.; Heberle, A.M.; Razquin Navas, P.; van Eunen, K.; Tolle, R.C.; Schwarz, J.J.; et al. A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat. Commun. 2016, 7, 13254. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Conn, C.S.; Qian, S.B. mTOR signaling in protein homeostasis: Less is more? Cell Cycle 2011, 10, 1940–1947. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell. Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatment Protocols. J. Chronic Fatigue Syndr. 2003, 11, 7–36. [Google Scholar] [CrossRef]
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.P.; Ngoei, K.R.; et al. Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech. 2016, 9, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Fox, J. The R commander: A basic-statistics graphical user interface to R. J. Stat. Softw. 2005, 14, 1–42. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J. Data Mining with Rattle and R; Use R! Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMS Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactome Pathway | Category | Dataset | Altered Fraction | Fold Enriched | Binomial Test p-Value |
|---|---|---|---|---|---|
| Gluconeogenesis (R-HSA-70263) | Carbohydrate metabolism | Proteomics | Upregulated | 3.81 | 0.022 |
| Pentose phosphate pathway (R-HSA-71336) | Carbohydrate metabolism | Proteomics | Upregulated | 4.52 | 0.03 |
| Citric acid (TCA) cycle and respiratory electron transport (R-HSA-1428517) | TCA cycle and respiration | Proteomics | Upregulated | 1.89 | 0.027 |
| Formation of ATP by chemiosmotic coupling (R-HSA-163210) | TCA cycle and respiration | Proteomics | Upregulated | 4.52 | 0.03 |
| Respiratory electron transport, ATP synthesis by chemiosmotic coupling, and heat production by uncoupling proteins. (R-HSA-163200) | TCA cycle and respiration | Transcriptomics | Downregulated | 3.59 | 1.33 × 10−11 |
| Respiratory electron transport (R-HSA-611105) | TCA cycle and respiration | Transcriptomics | Downregulated | 3.69 | 2.14 × 10−10 |
| Citric acid (TCA) cycle and respiratory electron transport (R-HSA-1428517) | TCA cycle and respiration | Transcriptomics | Downregulated | 2.83 | 1.95 × 10−9 |
| Complex I biogenesis (R-HSA-6799198) | TCA cycle and respiration | Transcriptomics | Downregulated | 4.19 | 7.59 × 10−8 |
| Beta-oxidation of lauroyl-CoA to decanoyl-CoA-CoA (R-HSA-77310) | Lipid metabolism | Proteomics | Upregulated | 13.57 | 0.0015 |
| Beta-oxidation of hexanoyl-CoA to butanoyl-CoA (R-HSA-77350) | Lipid metabolism | Proteomics | Upregulated | 10.85 | 0.0028 |
| Beta-oxidation of octanoyl-CoA to hexanoyl-CoA (R-HSA-77348) | Lipid metabolism | Proteomics | Upregulated | 10.85 | 0.0028 |
| Beta-oxidation of decanoyl-CoA to octanoyl-CoA-CoA (R-HSA-77346) | Lipid metabolism | Proteomics | Upregulated | 10.85 | 0.0028 |
| Mitochondrial fatty acid beta-oxidation of unsaturated fatty acids (R-HSA-77288) | Lipid metabolism | Proteomics | Upregulated | 10.85 | 0.0028 |
| Fatty acid metabolism (R-HSA-8978868) | Lipid metabolism | Proteomics | Upregulated | 2.87 | 0.0029 |
| Acyl chain remodeling of CL (R-HSA-1482798) | Lipid metabolism | Proteomics | Upregulated | 18.09 | 0.0057 |
| Beta-oxidation of myristoyl-CoA to lauroyl-CoA (R-HSA-77285) | Lipid metabolism | Proteomics | Upregulated | 18.09 | 0.0057 |
| Mitochondrial fatty acid beta-oxidation (R-HSA-77289) | Lipid metabolism | Proteomics | Upregulated | 5.51 | 3.32 × 10−4 |
| Beta-oxidation of palmitoyl-CoA to myristoyl-CoA (R-HSA-77305) | Lipid metabolism | Proteomics | Upregulated | 18.09 | 6.64 × 10−4 |
| Mitochondrial fatty acid beta-oxidation of saturated fatty acids (R-HSA-77286) | Lipid metabolism | Proteomics | Upregulated | 10.34 | 6.72 × 10−4 |
| NR1H2 and NR1H3 regulate gene expression linked to triglyceride lipolysis in adipose (R-HSA-9031528) | Lipid metabolism | Transcriptomics | Upregulated | 7.47 | 0.03 |
| Regulation of cholesterol biosynthesis by SREBP (SREBF) (R-HSA-1655829) | Lipid metabolism | Transcriptomics | Upregulated | 2.3 | 0.026 |
| Regulation of lipid metabolism by PPARalpha (R-HSA-400206) | Lipid metabolism | Transcriptomics | Upregulated | 1.87 | 0.031 |
| Phenylalanine and tyrosine metabolism (R-HSA-8963691) | Amino acid metabolism | Proteomics | Upregulated | 7.24 | 0.032 |
| Glutamate neurotransmitter release cycle (R-HSA-210500) | Amino acid metabolism | Proteomics | Upregulated | 13.57 | 0.0015 |
| Neurotransmitter release cycle (R-HSA-112310) | Amino acid metabolism | Proteomics | Upregulated | 6.03 | 0.014 |
| Metabolism of amino acids and derivatives (R-HSA-71291) | Amino acid metabolism | Proteomics | Upregulated | 1.64 | 0.021 |
| Aspartate and asparagine metabolism (R-HSA-8963693) | Amino acid metabolism | Proteomics | Upregulated | 7.24 | 0.032 |
| Lysine catabolism (R-HSA-71064) | Amino acid metabolism | Proteomics | Upregulated | 6.03 | 0.044 |
| Metabolism of amino acids and derivatives (R-HSA-71291) | Amino acid metabolism | Transcriptomics | Downregulated | 3.13 | 2.06 × 10−20 |
| Response of EIF2AK4 (GCN2) to amino acid deficiency (R-HSA-9633012) | Amino acid metabolism | Transcriptomics | Downregulated | 5.85 | 6.10 × 10−26 |
| Ubiquitin-dependent degradation of Cyclin D (R-HSA-75815) | Protein degradation | Transcriptomics | Downregulated | 2.93 | 2.75 × 10−4 |
| Autodegradation of the E3 ubiquitin ligase COP1 (R-HSA-349425) | Protein degradation | Transcriptomics | Downregulated | 2.93 | 2.75 × 10−4 |
| Vpu mediated degradation of CD4 (R-HSA-180534) | Protein degradation | Transcriptomics | Downregulated | 2.93 | 2.75 × 10−4 |
| Ubiquitin-mediated degradation of phosphorylated Cdc25A (R-HSA-69601) | Protein degradation | Transcriptomics | Downregulated | 2.93 | 2.75 × 10−4 |
| Degradation of GLI2 by the proteasome (R-HSA-5610783) | Protein degradation | Transcriptomics | Downregulated | 2.79 | 2.98 × 10−4 |
| GLI3 is processed to GLI3R by the proteasome (R-HSA-5610785) | Protein degradation | Transcriptomics | Downregulated | 2.79 | 2.98 × 10−4 |
| Degradation of GLI1 by the proteasome (R-HSA-5610780) | Protein degradation | Transcriptomics | Downregulated | 2.75 | 3.60 × 10−4 |
| Processing of SMDT1 (R-HSA-8949664) | Other mitochondrial | Proteomics | Upregulated | 6.78 | 0.01 |
| Release of apoptotic factors from the mitochondria (R-HSA-111457) | Other mitochondrial | Proteomics | Upregulated | 12.06 | 0.012 |
| Mitochondrial biogenesis (R-HSA-1592230) | Other mitochondrial | Proteomics | Upregulated | 2.81 | 0.013 |
| Transcriptional activation of mitochondrial biogenesis (R-HSA-2151201) | Other mitochondrial | Proteomics | Upregulated | 3.45 | 0.03 |
| Mitochondrial translation initiation (R-HSA-5368286) | Other mitochondrial | Transcriptomics | Downregulated | 3.37 | 1.80 × 10−8 |
| Mitochondrial translation termination (R-HSA-5419276) | Other mitochondrial | Transcriptomics | Downregulated | 3.37 | 1.80 × 10−8 |
| Mitochondrial translation (R-HSA-5368287) | Other mitochondrial | Transcriptomics | Downregulated | 3.26 | 2.23 × 10−8 |
| Mitochondrial protein import (R-HSA-1268020) | Other mitochondrial | Transcriptomics | Downregulated | 2.64 | 3.73 × 10−4 |
| Mitochondrial translation elongation (R-HSA-5389840) | Other mitochondrial | Transcriptomics | Downregulated | 3.48 | 5.03 × 10−9 |
| Mitochondrial calcium ion transport (R-HSA-8949215) | Other mitochondrial | Proteomics | Upregulated | 6.96 | 8.53 × 10−4 |
| Transport of nucleotide sugars (R-HSA-727802) | Transport of substrate molecules | Transcriptomics | Upregulated | 4.98 | 0.023 |
| SLC transporter disorders (R-HSA-5619102) | Transport of substrate molecules | Transcriptomics | Upregulated | 2.03 | 0.048 |
| Transport of small molecules (R-HSA-382551) | Transport of substrate molecules | Transcriptomics | Upregulated | 1.35 | 0.037 |
| Signaling by Leptin (R-HSA-2586552) | Metabolism | Proteomics | Upregulated | 9.05 | 0.021 |
| Diseases of metabolism (R-HSA-5668914) | Metabolism | Proteomics | Upregulated | 3.02 | 0.045 |
| Metabolism (R-HSA-1430728) | Metabolism | Proteomics | Upregulated | 1.51 | 2.12 × 10−4 |
| Activation of gene expression by SREBF (SREBP) (R-HSA-2426168) | Metabolism | Transcriptomics | Upregulated | 2.3 | 0.049 |
| Metabolism (R-HSA-1430728) | Metabolism | Transcriptomics | Downregulated | 1.43 | 3.80 × 10−8 |
| Functional Category | Number of Subunits Detected | Fraction Fold Change >1 in ME/CFS Proteomes | Binomial Test p Value | Mean Fold Change (± Standard Error) | Single-Sample t Test p Value |
|---|---|---|---|---|---|
| Complex I | 23 | 17/23 | 0.017 | 1.20 ± 0.05 | 3.44 × 10−4 |
| Complex II | 2 | 2/2 | NA | 1.08 ± 0.03 | NA |
| Complex III | 6 | 6/6 | 0.16 | 1.22 ± 0.03 | 7.69 × 10−4 |
| Complex IV | 10 | 7/10 | 0.17 | 1.07 ± 0.04 | 0.07 |
| Complex V | 22 | 18/22 | 2.17 × 10−3 | 1.11 ± 0.02 | 1.18 × 10−4 |
| All 5 Complexes | 63 | 50/63 | 1.51 × 10−6 | 1.14 ± 0.02 | 1.21 × 10−8 |
| TCA Cycle | 19 | 18/19 | 3.82 × 10−5 | 1.17 ± 0.04 | 1.03 × 10−4 |
| Protein import complex subunits | 11 | 4/11 | 0.89 | 1.00 ± 0.04 | 0.81 |
| SLC25 family | 11 | 9/11 | 0.033 | 1.33 ± 0.14 | 0.016 |
| Functional Category | Number of Transcripts Detected | Number of Transcripts with Fold Change <1 | Number of Transcripts with Fold Change >1 | Binomial Test p Value | Mean Fold Change (± Standard Error) | Single-Sample t Test p Value |
|---|---|---|---|---|---|---|
| Complex I | 39 | 37 | 2 | 2.84 × 10−9 | 0.81 ± 0.03 | 2.63 × 10−8 |
| Complex II | 4 | 0 | 4 | 0.13 | 1.12 ± 0.03 | 0.024 |
| Complex III | 9 | 8 | 1 | 0.039 | 0.82 ± 0.03 | 6.31 × 10−4 |
| Complex IV | 19 | 16 | 3 | 0.0044 | 0.83 ± 0.03 | 8.41 × 10−6 |
| Complex V | 18 | 17 | 1 | 1.45 × 10−4 | 0.80 ± 0.04 | 2.25 × 10−5 |
| All 5 complexes | 89 | 78 | 11 | 1.37 × 10−13 | 0.83 ± 0.02 | <2.2 × 10−16 |
| TCA cycle | 21 | 7 | 14 | 0.189 | 1.03 ± 0.02 | 0.171 |
| Protein import complex subunits | 25 | 20 | 5 | 0.0041 | 0.88 ± 0.02 | 3.29 × 10−5 |
| SLC25 family | 40 | 21 | 19 | 0.88 | 1.011 ± 0.02 | 0.628 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Missailidis, D.; Sanislav, O.; Allan, C.Y.; Smith, P.K.; Annesley, S.J.; Fisher, P.R. Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts. Int. J. Mol. Sci. 2021, 22, 2046. https://doi.org/10.3390/ijms22042046
Missailidis D, Sanislav O, Allan CY, Smith PK, Annesley SJ, Fisher PR. Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts. International Journal of Molecular Sciences. 2021; 22(4):2046. https://doi.org/10.3390/ijms22042046
Chicago/Turabian StyleMissailidis, Daniel, Oana Sanislav, Claire Y. Allan, Paige K. Smith, Sarah J. Annesley, and Paul R. Fisher. 2021. "Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts" International Journal of Molecular Sciences 22, no. 4: 2046. https://doi.org/10.3390/ijms22042046
APA StyleMissailidis, D., Sanislav, O., Allan, C. Y., Smith, P. K., Annesley, S. J., & Fisher, P. R. (2021). Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts. International Journal of Molecular Sciences, 22(4), 2046. https://doi.org/10.3390/ijms22042046