
Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions

 ,
, 
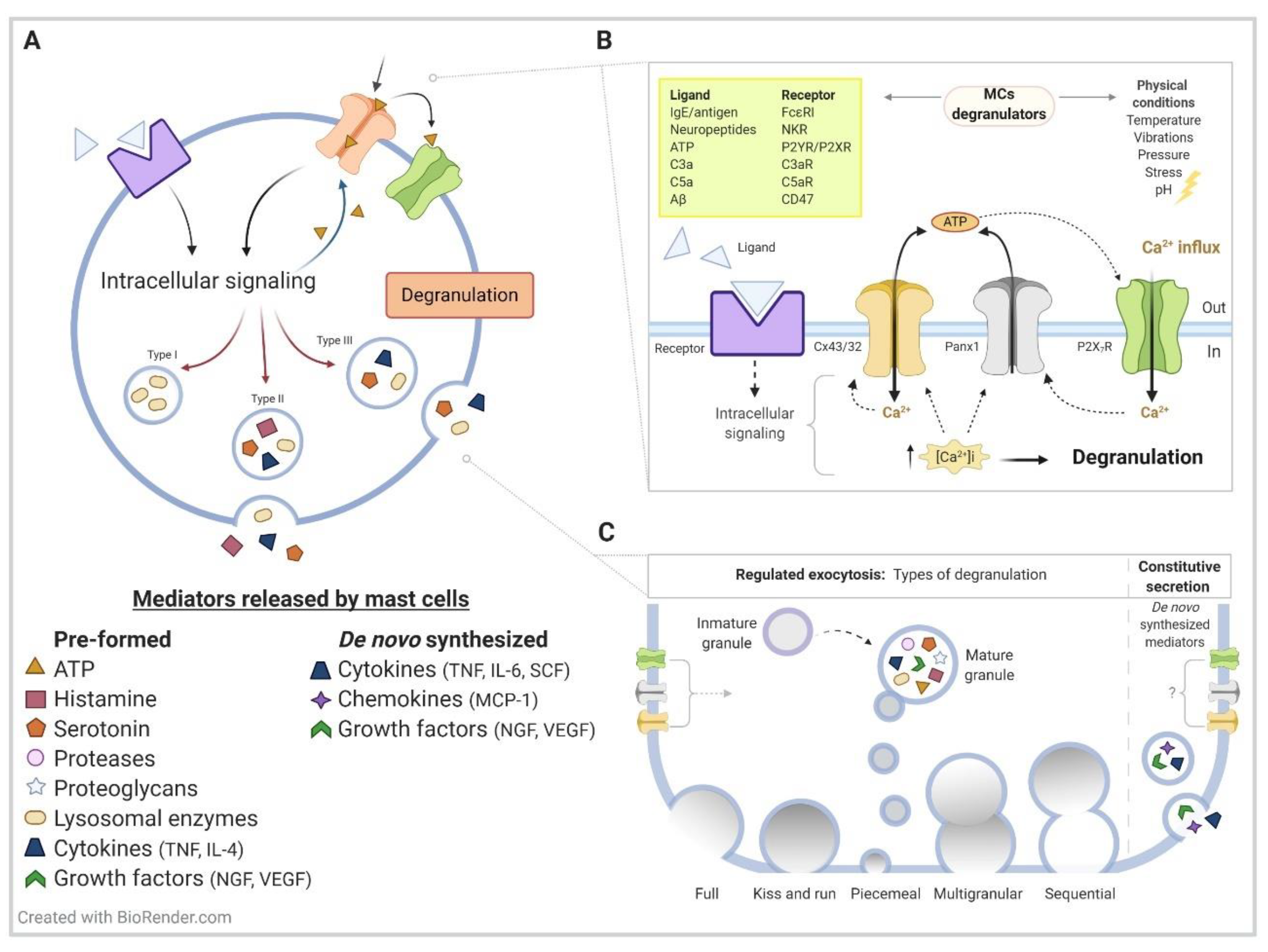{kind=link}
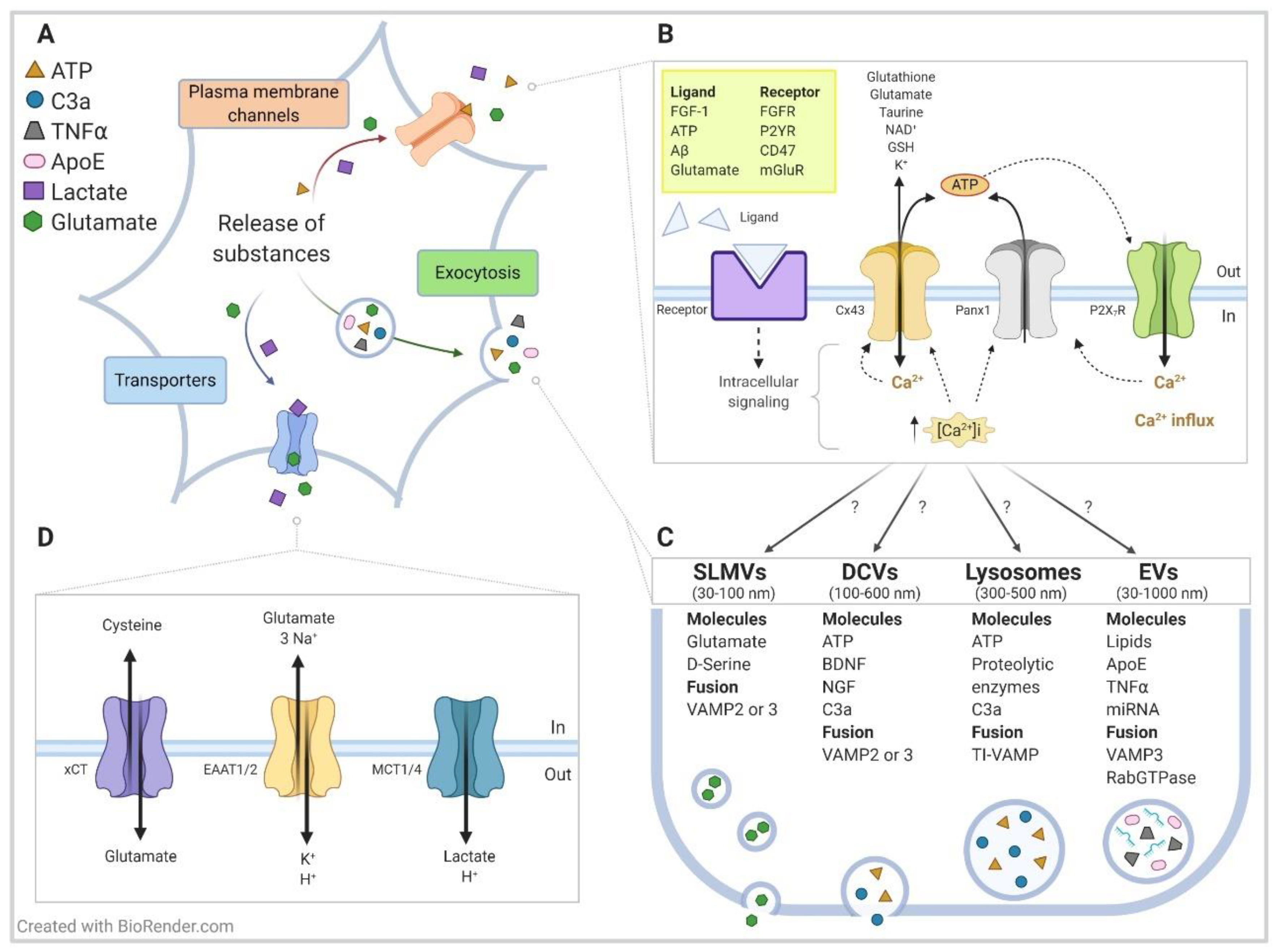{kind=link}
Abstract
1. Mast Cell Generalities
2. Hemichannels Overview
3. Hemichannels on Mast Cell Immunological Function
4. Contribution of Mast Cells Hemichannels on Neuroinflammatory Conditions
4.1. Alzheimer’s Disease
4.2. Harmful Stress Conditions
4.3. Amyotrophic Lateral Sclerosis
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dropp, J.J. Mast cells in the central nervous system of several rodents. Anat. Rec. 1972, 174, 227–237. [Google Scholar] [CrossRef]
- Ibrahim, M.Z.M. The mast cells of the mammalian central nervous system. J. Neurol. Sci. 1974, 21, 479–499. [Google Scholar] [CrossRef]
- Krüger, P.G. Demonstration of mast cells in the albino rat brain. Experientia 1974, 30, 810–811. [Google Scholar] [CrossRef] [PubMed]
- Dropp, J.J. Mast cells in mammalian brain. Acta Anat. 1976, 94, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C. Mast cells: The immune gate to the brain. Life Sci. 1990, 46, 607–617. [Google Scholar] [CrossRef]
- Crivellato, E.; Beltrami, C.A.; Mallardi, F.; Ribatti, D. Paul Ehrlich’s doctoral thesis: A milestone in the study of mast cells. Br. J. Haematol. 2003, 123, 19–21. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef]
- Dudeck, A.; Köberle, M.; Goldmann, O.; Meyer, N.; Dudeck, J.; Lemmens, S.; Rohde, M.; Roldán, N.G.; Dietze-Schwonberg, K.; Orinska, Z.; et al. Mast cells as protectors of health. J. Allergy Clin. Immunol. 2019, 144, S4–S18. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadou, V.; Lambracht-Hall, M.; Reichler, J.; Theoharides, T.C. Histochemical and ultrastructural characteristics of rat brain perivascular mast cells stimulated with compound 48/80 and carbachol. Neuroscience 1990, 39, 209–224. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Redegeld, F.A.; Yu, Y.; Kumari, S.; Charles, N.; Blank, U. Non-IgE mediated mast cell activation. Immunol. Rev. 2018, 282, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I.; Ren, H. Recent advances in our understanding of mast cell activation—Or should it be mast cell mediator disorders? Expert Rev. Clin. Immunol. 2019, 15, 639–656. [Google Scholar] [CrossRef]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast Cell Mediators: Their Differential Release and the Secretory Pathways Involved. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Xu, H.; Bin, N.-R.; Sugita, S. Diverse exocytic pathways for mast cell mediators. Biochem. Soc. Trans. 2018, 46, 235–247. [Google Scholar] [CrossRef]
- Blank, U. The mechanisms of exocytosis in mast cells. Adv. Exp. Med. Biol. 2011, 716, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Sagi-Eisenberg, R. Anaphylactic Degranulation of Mast Cells: Focus on Compound Exocytosis. J. Immunol. Res. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Raposo, G.; Tenza, D.; Mecheri, S.; Peronet, R.; Bonnerot, C.; Desaymard, C. Accumulation of Major Histocompatibility Complex Class II Molecules in Mast Cell Secretory Granules and Their Release upon Degranulation. Mol. Biol. Cell 1997, 8, 2631–2645. [Google Scholar] [CrossRef]
- Dvorak, A.M. Ultrastructural Studies of Human Basophils and Mast Cells. J. Histochem. Cytochem. 2005, 53, 1043–1070. [Google Scholar] [CrossRef]
- Puri, N.; Roche, P.A. Mast cells possess distinct secretory granule subsets whose exocytosis is regulated by different SNARE isoforms. Proc. Natl. Acad. Sci. USA 2008, 105, 2580–2585. [Google Scholar] [CrossRef]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Lorentz, A.; Baumann, A.; Vitte, J.; Blank, U. The SNARE Machinery in Mast Cell Secretion. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Madera-Salcedo, I.K.; Danelli, L.; Claver, J.; Tiwari, N.; Sãnchez-Miranda, E.; Vãzquez-Victorio, G.; Ramírez-Valadez, K.A.; Macias-Silva, M.; Gonzãlez-Espinosa, C. Vesicular Trafficking and Signaling for Cytokine and Chemokine Secretion in Mast Cells. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Valitutti, S. New roles and controls of mast cells. Curr. Opin. Immunol. 2018, 50, 39–47. [Google Scholar] [CrossRef]
- Harcha, P.A.; López, X.; Sáez, P.J.; Fernández, P.; Barría, I.; Martínez, A.D.; Sáez, J.C. Pannexin-1 Channels Are Essential for Mast Cell Degranulation Triggered During Type I Hypersensitivity Reactions. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Harcha, P.A.; Vargas, R.A.; Yi, C.; Koulakoff, A.; Giaume, C.; Saez, J.C. Hemichannels Are Required for Amyloid -Peptide-Induced Degranulation and Are Activated in Brain Mast Cells of APPswe/PS1dE9 Mice. J. Neurosci. 2015, 35, 9526–9538. [Google Scholar] [CrossRef][Green Version]
- Ogasawara, T.; Murakami, M.; Suzuki-Nishimura, T.; Uchida, M.K.; Kudo, I. Mouse bone marrow-derived mast cells undergo exocytosis, prostanoid generation, and cytokine expression in response to G protein-activating polybasic compounds after coculture with fibroblasts in the presence of c-kit ligand. J. Immunol. 1997, 158, 393–404. [Google Scholar] [PubMed]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef]
- Karimi, K.; Kool, M.; Nijkamp, F.P.; Redegeld, F.A. Substance P can stimulate prostaglandin D2 and leukotriene C4 generation without granule exocytosis in murine mast cells. Eur. J. Pharmacol. 2004, 489, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Reynier-Rebuffel, A.M.; Mathiau, P.; Callebert, J.; Dimitriadou, V.; Farjaudon, N.; Kacem, K.; Launay, J.M.; Seylaz, J.; Abineau, P. Substance P, calcitonin gene-related peptide, and capsaicin release serotonin from cerebrovascular mast cells. Am. J. Physiol. Integr. Comp. Physiol. 1994, 267, R1421–R1429. [Google Scholar] [CrossRef]
- Kulka, M.; Sheen, C.H.; Tancowny, B.P.; Grammer, L.C.; Schleimer, R.P. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology 2008, 123, 398–410. [Google Scholar] [CrossRef]
- Lewis, R.A.; Soter, N.A.; Diamond, P.T.; Austen, K.F.; Oates, J.A.; Roberts, L.J. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J. Immunol. 1982, 129, 1627–1631. [Google Scholar]
- Burd, P.R.; Rogers, H.W.; Gordon, J.R.; Martin, C.A.; Jayaraman, S.; Wilson, S.D.; Dvorak, A.M.; Galli, S.J.; Dorf, M.E. Interleukin 3-dependent and -independent mast cells stimulated with IgE and antigen express multiple cytokines. J. Exp. Med. 1989, 170, 245–257. [Google Scholar] [CrossRef]
- MacDonald, A.J.; Pick, J.; Bissonnette, E.Y.; Befus, A.D. Rat mucosal mast cells: The cultured bone marrow-derived mast cell is biochemically and functionally analogous to its counterpart in vivo. Immunology 1998, 93, 533–539. [Google Scholar] [CrossRef]
- Gilchrist, M.; McCauley, S.D.; Befus, A.D. Expression, localization, and regulation of NOS in human mast cell lines: Effects on leukotriene production. Blood 2004, 104, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Swindle, E.J.; Metcalfe, D.D.; Coleman, J.W. Rodent and Human Mast Cells Produce Functionally Significant Intracellular Reactive Oxygen Species but Not Nitric Oxide. J. Biol. Chem. 2004, 279, 48751–48759. [Google Scholar] [CrossRef]
- Wang, X.; Sam, S.W.; Yip, K.H.; Lau, H.Y.A. Functional characterization of human mast cells cultured from adult peripheral blood. Int. Immunopharmacol. 2006, 6, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Yoshimura, T.; Parsons, T.; Befus, A.D. Microenvironmental regulation of inducible nitric oxide synthase expression and nitric oxide production in mouse bone marrow-derived mast cells. J. Leukoc. Biol. 2012, 91, 581–590. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, J.D.; Lin, T.J.; Marshall, J.S. Toll-like receptor 4-mediated activation of murine mast cells. J. Leukoc. Biol. 2001, 70, 977–984. [Google Scholar] [PubMed]
- King, C.A.; Marshall, J.S.; Alshurafa, H.; Anderson, R. Release of Vasoactive Cytokines by Antibody-Enhanced Dengue Virus Infection of a Human Mast Cell/Basophil Line. J. Virol. 2000, 74, 7146–7150. [Google Scholar] [CrossRef]
- Kulka, M.; Alexopoulou, L.; Flavell, R.A.; Metcalfe, D.D. Activation of mast cells by double-stranded RNA: Evidence for activation through Toll-like receptor 3. J. Allergy Clin. Immunol. 2004, 114, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, J.B.; Hair, G.A.; Ansari, A.A.; Secor, W.E.; Gilfillan, A.M.; Metcalfe, D.D.; Kirshenbaum, A.S. IgE-FcεRI Interactions Determine HIV Coreceptor Usage and Susceptibility to Infection during Ontogeny of Mast Cells. J. Immunol. 2009, 182, 6401–6409. [Google Scholar] [CrossRef] [PubMed]
- El-Lati, S.G.; Church, M.K.; Dahinden, C.A. Complement Peptides C3a- and C5a-Induced Mediator Release from Dissociated Human Skin Mast Cells. J. Investig. Dermatol. 1994, 102, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Henz, B.M.; Krüger-Krasagakes, S.; Köhl, J.; Burger, R.; Guhl, S.; Haase, I.; Lippert, U.; Zuberbier, T. C3a and C5a stimulate chemotaxis of human mast cells. Blood 1997, 89, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Erdei, A.; Márton, A.; Hajna, P.; Gábor, T.; Israel, P. Regulation of mast cell activation by complement-derived peptides. Immunol. Lett. 2004, 92, 39–42. [Google Scholar] [CrossRef]
- Esseltine, J.L.; Laird, D.W. Next-Generation Connexin and Pannexin Cell Biology. Trends Cell Biol. 2016, 26, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta 2018, 1860, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Naus, C.C.; Sáez, J.C.; Leybaert, L. Glial Connexins and Pannexins in the Healthy and Diseased Brain. Physiol. Rev. 2021, 101, 93–145. [Google Scholar] [CrossRef]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular Calcium Signaling in Astrocytes via ATP Release through Connexin Hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef]
- Bruzzone, R.; Barbe, M.T.; Jakob, N.J.; Monyer, H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J. Neurochem. 2005, 92, 1033–1043. [Google Scholar] [CrossRef]
- Cherian, P.P.; Siller-Jackson, A.J.; Gu, S.; Wang, X.; Bonewald, L.F.; Sprague, E.; Jiang, J.X. Mechanical Strain Opens Connexin 43 Hemichannels in Osteocytes: A Novel Mechanism for the Release of Prostaglandin. Mol. Biol. Cell 2005, 16, 3100–3106. [Google Scholar] [CrossRef]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 Hemichannels and Gap Junction Channels in Astrocytes Are Regulated Oppositely by Proinflammatory Cytokines Released from Activated Microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Cea, L.A.; Vega, J.L.; Boric, M.P.; Monyer, H.; Bennett, M.V.L.; Frank, M.; Willecke, K.; Sáez, J.C. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 2013, 75, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A. Role of Connexin/Pannexin containing channels in infectious diseases. FEBS Lett. 2014, 588, 1389–1395. [Google Scholar] [CrossRef]
- Eugenin, E.A. Role of cell-to-cell communication in cancer: New features, insights, and directions. Cancer Rep. 2019, 2. [Google Scholar] [CrossRef]
- Sáez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Panchina, Y.; Kelmanson, I.; Matz, M.; Lukyanov, K.; Usman, N.; Lukyanov, S. A ubiquitous family of putative gap junction molecules. Curr. Biol. 2000, 10, R473–R474. [Google Scholar] [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef]
- Locovei, S.; Wang, J.; Dahl, G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006, 580, 239–244. [Google Scholar] [CrossRef]
- Locovei, S.; Scemes, E.; Qiu, F.; Spray, D.C.; Dahl, G. Pannexin1 is part of the pore forming unit of the P2X 7 receptor death complex. FEBS Lett. 2007, 581, 483–488. [Google Scholar] [CrossRef]
- Qiu, F.; Dahl, G. A permeant regulating its permeation pore: Inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Physiol. 2009, 296, C250–C255. [Google Scholar] [CrossRef]
- Kurtenbach, S.; Prochnow, N.; Kurtenbach, S.; Klooster, J.; Zoidl, C.; Dermietzel, R.; Kamermans, M.; Zoidl, G. Pannexin1 Channel Proteins in the Zebrafish Retina Have Shared and Unique Properties. PLoS ONE 2013, 8, e77722. [Google Scholar] [CrossRef]
- López, X.; Escamilla, R.; Fernández, P.; Duarte, Y.; González-Nilo, F.; Palacios-Prado, N.; Martinez, A.D.; Sáez, J.C. Stretch-Induced Activation of Pannexin 1 Channels Can Be Prevented by PKA-Dependent Phosphorylation. Int. J. Mol. Sci. 2020, 21, 9180. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.J.; Zhou, N.; MacVicar, B.A. Ischemia Opens Neuronal Gap Junction Hemichannels. Science 2006, 312, 924–927. [Google Scholar] [CrossRef]
- Iglesias, R.; Locovei, S.; Roque, A.; Alberto, A.P.; Dahl, G.; Spray, D.C.; Scemes, E. P2X 7 receptor-Pannexin1 complex: Pharmacology and signaling. Am. J. Physiol. Physiol. 2008, 295, C752–C760. [Google Scholar] [CrossRef] [PubMed]
- Bunse, S.; Locovei, S.; Schmidt, M.; Qiu, F.; Zoidl, G.; Dahl, G.; Dermietzel, R. The potassium channel subunit Kvβ3 interacts with pannexin 1 and attenuates its sensitivity to changes in redox potentials. FEBS J. 2009, 276, 6258–6270. [Google Scholar] [CrossRef]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Saez, P.J.; Jiang, J.X.; Naus, C.C.; Saez, J.C.; Giaume, C. Amyloid -Induced Death in Neurons Involves Glial and Neuronal Hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Weilinger, N.L.; Tang, P.L.; Thompson, R.J. Anoxia-Induced NMDA Receptor Activation Opens Pannexin Channels via Src Family Kinases. J. Neurosci. 2012, 32, 12579–12588. [Google Scholar] [CrossRef]
- Weilinger, N.L.; Maslieieva, V.; Bialecki, J.; Sridharan, S.S.; Tang, P.L.; Thompson, R.J. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol. Sin. 2013, 34, 39–48. [Google Scholar] [CrossRef]
- Beckel, J.M.; Argall, A.J.; Lim, J.C.; Xia, J.; Lu, W.; Coffey, E.E.; Macarak, E.J.; Shahidullah, M.; Delamere, N.A.; Zode, G.S.; et al. Mechanosensitive release of adenosine 5′-triphosphate through pannexin channels and mechanosensitive upregulation of pannexin channels in optic nerve head astrocytes: A mechanism for purinergic involvement in chronic strain. Glia 2014, 62, 1486–1501. [Google Scholar] [CrossRef] [PubMed]
- Olivier, E.; Dutot, M.; Regazzetti, A.; Leguillier, T.; Dargère, D.; Auzeil, N.; Laprévote, O.; Rat, P. P2X7-pannexin-1 and amyloid β-induced oxysterol input in human retinal cell: Role in age-related macular degeneration? Biochimie 2016, 127, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Wang, J.; Dahl, G. Alanine substitution scanning of pannexin1 reveals amino acid residues mediating ATP sensitivity. Purinergic Signal. 2012, 8, 81–90. [Google Scholar] [CrossRef]
- Boyce, A.K.J.; Swayne, L.A. P2X7 receptor cross-talk regulates ATP-induced pannexin 1 internalization. Biochem. J. 2017, 474, 2133–2144. [Google Scholar] [CrossRef]
- Ma, H.-T.; Beaven, M.A. Regulation of Ca2+ Signaling with Particular Focus on Mast Cells. Crit. Rev. Immunol. 2009, 29, 155–186. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Beaven, M.A. Regulation of Mast Cell Responses in Health and Disease. Crit. Rev. Immunol. 2011, 31, 475–530. [Google Scholar] [CrossRef] [PubMed]
- Maldifassi, M.C.; Momboisse, F.; Guerra, M.J.; Vielma, A.H.; Maripillán, J.; Báez-Matus, X.; Flores-Muñoz, C.; Cádiz, B.; Schmachtenberg, O.; Martínez, A.D.; et al. The interplay between α7 nicotinic acetylcholine receptors, pannexin-1 channels and P2X7 receptors elicit exocytosis in chromaffin cells. J. Neurochem. 2020, jnc.15186. [Google Scholar] [CrossRef]
- Sudheer, P.S.; Hall, J.E.; Donev, R.; Read, G.; Rowbottom, A.; Williams, P.E. Nicotinic acetylcholine receptors on basophils and mast cells. Anaesthesia 2006, 61, 1170–1174. [Google Scholar] [CrossRef]
- Kageyama-Yahara, N.; Suehiro, Y.; Yamamoto, T.; Kadowaki, M. IgE-induced degranulation of mucosal mast cells is negatively regulated via nicotinic acetylcholine receptors. Biochem. Biophys. Res. Commun. 2008, 377, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shi, X.; Li, X.; Zou, J.; Zhou, C.; Liu, W.; Shao, H.; Chen, H.; Shi, L. Neurotransmitter and neuropeptide regulation of mast cell function: A systematic review. J. Neuroinflamm. 2020, 17, 356. [Google Scholar] [CrossRef]
- Vliagoftis, H.; Hutson, A.M.; Mahmudi-Azer, S.; Kim, H.; Rumsaeng, V.; Oh, C.K.; Moqbel, R.; Metcalfe, D.D. Mast cells express connexins on their cytoplasmic membrane. J. Allergy Clin. Immunol. 1999, 103, 656–662. [Google Scholar] [CrossRef]
- Oliani, S.M.; Giro, A.P.; Smith, R.L. Gap Junctions between Mast Cells and Fibroblasts in the Developing Avian Eye. Cells Tissues Organs 1995, 154, 267–271. [Google Scholar] [CrossRef]
- Moyer, K.E.; Saggers, G.C.; Ehrlich, H.P. Mast cells promote fibroblast populated collagen lattice contraction through gap junction intercellular communication. Wound Repair Regen. 2004, 12, 269–275. [Google Scholar] [CrossRef]
- Pistorio, A.L.; Ehrlich, H.P. Modulatory effects of connexin-43 expression on gap junction intercellular communications with mast cells and fibroblasts. J. Cell. Biochem. 2011, 112, 1441–1449. [Google Scholar] [CrossRef]
- Koulakoff, A.; Mei, X.; Orellana, J.A.; Sáez, J.C.; Giaume, C. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1818, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Suzumura, A. Gap junctions and hemichannels composed of connexins: Potential therapeutic targets for neurodegenerative diseases. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Gajardo-Gómez, R.; Labra, V.C.; Orellana, J.A. Connexins and Pannexins: New Insights into Microglial Functions and Dysfunctions. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef]
- Yi, C.; Koulakoff, A.; Giaume, C. Astroglial Connexins as a Therapeutic Target for Alzheimer’s Disease. Curr. Pharm. Des. 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Yang, T.; Cui, S.; Chen, G. Connexin Hemichannels in Astrocytes: Role in CNS Disorders. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef]
- Nagy, J.I.; Li, W.; Hertzberg, E.L.; Marotta, C.A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Mei, X.; Ezan, P.; Giaume, C.; Koulakoff, A. Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 2010, 171, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Mizoguchi, H.; Doi, Y.; Jin, S.; Noda, M.; Liang, J.; Li, H.; Zhou, Y.; Mori, R.; Yasuoka, S.; et al. Blockade of Gap Junction Hemichannel Suppresses Disease Progression in Mouse Models of Amyotrophic Lateral Sclerosis and Alzheimer’s Disease. PLoS ONE 2011, 6, e21108. [Google Scholar] [CrossRef]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Roux, L.; Dallérac, G.; Matias, I.; Dulong, J.; Mothet, J.P.; Rouach, N.; Giaume, C. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia 2015, 63, 795–811. [Google Scholar] [CrossRef]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef]
- Maslinska, D.; Laure-Kamionowska, M.; Maslinski, K.T.; Gujski, M.; Maslinski, S. Distribution of tryptase-containing mast cells and metallothionein reactive astrocytes in human brains with amyloid deposits. Inflamm. Res. 2007, 56, S17–S18. [Google Scholar] [CrossRef] [PubMed]
- Kvetnoĭ, I.M.; Kvetnaia, T.V.; Riadnova, I.I.; Fursov, B.B.; Ernandes-Jago, H.; Blesa, J.R. Expression of beta-amyloid and tau-protein in mastocytes in Alzheimer disease. Arkh. Patol. 2003, 65, 36–39. [Google Scholar] [PubMed]
- Niederhoffer, N.; Levy, R.; Sick, E.; Andre, P.; Coupin, G.; Lombard, Y.; Gies, J.-P. Amyloid β Peptides Trigger CD47-Dependent Mast Cell Secretory and Phagocytic Responses. Int. J. Immunopathol. Pharmacol. 2009, 22, 473–483. [Google Scholar] [CrossRef]
- Sick, E.; Niederhoffer, N.; Takeda, K.; Landry, Y.; Gies, J.-P. Activation of CD47 receptors causes histamine secretion from mast cells. Cell. Mol. Life Sci. 2009, 66, 1271–1282. [Google Scholar] [CrossRef]
- Liang, J.; Sloane, J.A.; Wells, J.M.; Abraham, C.R.; Fine, R.E.; Sipe, J.D. Evidence for local production of acute phase response apolipoprotein serum amyloid A in Alzheimer’s disease brain. Neurosci. Lett. 1997, 225, 73–76. [Google Scholar] [CrossRef]
- Guo, J.; Yu, J.; Grass, D.; de Beer, F.C.; Kindy, M.S. Inflammation-Dependent Cerebral Deposition of Serum Amyloid A Protein in a Mouse Model of Amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef]
- Olsson, N.; Siegbahn, A.; Nilsson, G. Serum Amyloid A Induces Chemotaxis of Human Mast Cells by Activating a Pertussis Toxin-Sensitive Signal Transduction Pathway. Biochem. Biophys. Res. Commun. 1999, 254, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Niemi, K.; Baumann, M.H.; Kovanen, P.T.; Eklund, K.K. Serum amyloid A (SAA) activates human mast cells which leads into degradation of SAA and generation of an amyloidogenic SAA fragment. Biochim. Biophys. Acta 2006, 1762, 424–430. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, W.; Xu, L.; Pan, J.; Zheng, X.; Chen, Z. Effect of cerebral ischemia on brain mast cells in rats. Brain Res. 2004, 1019, 275–280. [Google Scholar] [CrossRef]
- Biran, V.; Cochois, V.; Karroubi, A.; Arrang, J.M.; Charriaut-Marlangue, C.; Héron, A. Stroke Induces Histamine Accumulation and Mast Cell Degranulation in the Neonatal Rat Brain. Brain Pathol. 2008, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mattila, O.S.; Strbian, D.; Saksi, J.; Pikkarainen, T.O.; Rantanen, V.; Tatlisumak, T.; Lindsberg, P.J. Cerebral Mast Cells Mediate Blood-Brain Barrier Disruption in Acute Experimental Ischemic Stroke Through Perivascular Gelatinase Activation. Stroke 2011, 42, 3600–3605. [Google Scholar] [CrossRef]
- Strbian, D.; Karjalainen-Lindsberg, M.-L.; Kovanen, P.T.; Tatlisumak, T.; Lindsberg, P.J. Mast Cell Stabilization Reduces Hemorrhage Formation and Mortality After Administration of Thrombolytics in Experimental Ischemic Stroke. Circulation 2007, 116, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Silverman, A.-J.; Vannucci, S.J. Mast Cell Stabilization Limits Hypoxic-Ischemic Brain Damage in the Immature Rat. Dev. Neurosci. 2007, 29, 373–384. [Google Scholar] [CrossRef]
- Lozada, A.; Maegele, M.; Stark, H.; Neugebauer, E.M.A.; Panula, P. Traumatic brain injury results in mast cell increase and changes in regulation of central histamine receptors. Neuropathol. Appl. Neurobiol. 2005, 31, 150–162. [Google Scholar] [CrossRef]
- Stokely, M.E.; Orr, E.L. Acute Effects of Calvarial Damage on Dural Mast Cells, Pial Vascular Permeability, and Cerebral Cortical Histamine Levels in Rats and Mice. J. Neurotrauma 2008, 25, 52–61. [Google Scholar] [CrossRef]
- Sayed, B.A.; Christy, A.L.; Walker, M.E.; Brown, M.A. Meningeal Mast Cells Affect Early T Cell Central Nervous System Infiltration and Blood-Brain Barrier Integrity through TNF: A Role for Neutrophil Recruitment? J. Immunol. 2010, 184, 6891–6900. [Google Scholar] [CrossRef] [PubMed]
- Pinke, K.; Zorzella-Pezavento, S.G.; Lara, V.; Sartori, A. Should mast cells be considered therapeutic targets in multiple sclerosis? Neural Regen. Res. 2020, 15, 1995. [Google Scholar] [CrossRef] [PubMed]
- Esposito, P.; Gheorghe, D.; Kandere, K.; Pang, X.; Connolly, R.; Jacobson, S.; Theoharides, T.C. Acute stress increases permeability of the blood–brain-barrier through activation of brain mast cells. Brain Res. 2001, 888, 117–127. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Li, T.; Martin, E.; Abada, Y.; Boucher, C.; Cès, A.; Youssef, I.; Fenaux, G.; Forand, Y.; Legrand, A.; Nachiket, N.; et al. Effects of Chronic Masitinib Treatment in APPswe/PSEN1dE9 Transgenic Mice Modeling Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 1339–1345. [Google Scholar] [CrossRef]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.-P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimer’s Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Benrabah, R.; Schmidt, N.; Zéphir, H.; Clavelou, P.; Vongsouthi, C.; Dubreuil, P.; Moussy, A.; Hermine, O. Masitinib treatment in patients with progressive multiple sclerosis: A randomized pilot study. BMC Neurol. 2012, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedrós, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Spanos, C.; Pang, X.; Alferes, L.; Ligris, K.; Letourneau, R.; Rozniecki, J.J.; Webster, E.; Chrousos, G.P. Stress-induced intracranial mast cell degranulation: A corticotropin-releasing hormone-mediated effect. Endocrinology 1995, 136, 5745–5750. [Google Scholar] [CrossRef]
- Wang, W.; Ji, P.; Dow, K.E. Corticotropin-releasing hormone induces proliferation and TNF-α release in cultured rat microglia via MAP kinase signalling pathways. J. Neurochem. 2002, 84, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Papadopoulou, N.; Kempuraj, D.; Boucher, W.S.; Sugimoto, K.; Cetrulo, C.L.; Theoharides, T.C. Human Mast Cells Express Corticotropin-Releasing Hormone (CRH) Receptors and CRH Leads to Selective Secretion of Vascular Endothelial Growth Factor. J. Immunol. 2005, 174, 7665–7675. [Google Scholar] [CrossRef]
- Ryu, J.K.; Cho, T.; Choi, H.B.; Wang, Y.T.; McLarnon, J.G. Microglial VEGF Receptor Response Is an Integral Chemotactic Component in Alzheimer’s Disease Pathology. J. Neurosci. 2009, 29, 3–13. [Google Scholar] [CrossRef]
- Karagkouni, A.; Alevizos, M.; Theoharides, T.C. Effect of stress on brain inflammation and multiple sclerosis. Autoimmun. Rev. 2013, 12, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Kritas, S.K.; Saggini, A.; Cerulli, G.; Caraffa, A.; Antinolfi, P.; Pantalone, A.; Rosati, M.; Tei, M.; Speziali, A.; Saggini, R.; et al. Corticotropin-Releasing Hormone, Microglia and Mental Disorders. Int. J. Immunopathol. Pharmacol. 2014, 27, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Orellana, J.A.; Moraga-Amaro, R.; DÃ-az-Galarce, R.; Rojas, S.; Maturana, C.J.; Stehberg, J.; Sáez, J.C. Restraint stress increases hemichannel activity in hippocampal glial cells and neurons. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Maturana, C.J.; Aguirre, A.; Sáez, J.C. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev. Neurobiol. 2017, 77, 625–642. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease: Table 1. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Fritz, E.; Izaurieta, P.; Weiss, A.; Mir, F.R.; Rojas, P.; Gonzalez, D.; Rojas, F.; Brown, R.H.; Madrid, R.; van Zundert, B. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J. Neurophysiol. 2013, 109, 2803–2814. [Google Scholar] [CrossRef]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis Drives Motor Neuron Death in Models of Both Sporadic and Familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Rojas, F.; Cortes, N.; Abarzua, S.; Dyrda, A.; van Zundert, B. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Ikiz, B.; Alvarez, M.J.; Ré, D.B.; Le Verche, V.; Politi, K.; Lotti, F.; Phani, S.; Pradhan, R.; Yu, C.; Croft, G.F.; et al. The Regulatory Machinery of Neurodegeneration in In Vitro Models of Amyotrophic Lateral Sclerosis. Cell Rep. 2015, 12, 335–345. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 2019, 40, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Jury, N.; Abarzua, S.; Diaz, I.; Guerra, M.V.; Ampuero, E.; Cubillos, P.; Martinez, P.; Herrera-Soto, A.; Arredondo, C.; Rojas, F.; et al. Widespread loss of the silencing epigenetic mark H3K9me3 in astrocytes and neurons along with hippocampal-dependent cognitive impairment in C9orf72 BAC transgenic mice. Clin. Epigenet. 2020, 12, 32. [Google Scholar] [CrossRef]
- Mishra, V.; Re, D.B.; Le Verche, V.; Alvarez, M.J.; Vasciaveo, A.; Jacquier, A.; Doulias, P.-T.; Greco, T.M.; Nizzardo, M.; Papadimitriou, D.; et al. Systematic elucidation of neuron-astrocyte interaction in models of amyotrophic lateral sclerosis using multi-modal integrated bioinformatics workflow. Nat. Commun. 2020, 11, 5579. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Vinken, M.; Rogiers, V.; Bukauskas, F.F.; Bultynck, G.; Leybaert, L. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta Biomembr. 2013, 1828, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Garre, J.M.; Yang, G.; Bukauskas, F.F.; Bennett, M.V.L. FGF-1 Triggers Pannexin-1 Hemichannel Opening in Spinal Astrocytes of Rodents and Promotes Inflammatory Responses in Acute Spinal Cord Slices. J. Neurosci. 2016, 36, 4785–4801. [Google Scholar] [CrossRef]
- Hong, Y.; Zhao, T.; Li, X.-J.; Li, S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef]
- Varela-Eirin, M.; Varela-Vazquez, A.; Rodríguez-Candela Mateos, M.; Vila-Sanjurjo, A.; Fonseca, E.; Mascareñas, J.L.; Eugenio Vázquez, M.; Mayan, M.D. Recruitment of RNA molecules by connexin RNA-binding motifs: Implication in RNA and DNA transport through microvesicles and exosomes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 728–736. [Google Scholar] [CrossRef]
- Cotrina, M.L.; Gao, Q.; Lin, J.H.-C.; Nedergaard, M. Expression and function of astrocytic gap junctions in aging. Brain Res. 2001, 901, 55–61. [Google Scholar] [CrossRef]
- Rochefort, N.; Quenech’du, N.; Ezan, P.; Giaume, C.; Milleret, C. Postnatal development of GFAP, connexin43 and connexin30 in cat visual cortex. Dev. Brain Res. 2005, 160, 252–264. [Google Scholar] [CrossRef]
- Diaz-Amarilla, P.; Olivera-Bravo, S.; Trias, E.; Cragnolini, A.; Martinez-Palma, L.; Cassina, P.; Beckman, J.; Barbeito, L. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 18126–18131. [Google Scholar] [CrossRef]
- Keller, A.F.; Gravel, M.; Kriz, J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp. Neurol. 2011, 228, 69–79. [Google Scholar] [CrossRef]
- Cui, Y.; Masaki, K.; Yamasaki, R.; Imamura, S.; Suzuki, S.O.; Hayashi, S.; Sato, S.; Nagara, Y.; Kawamura, M.F.; Kira, J. Extensive dysregulations of oligodendrocytic and astrocytic connexins are associated with disease progression in an amyotrophic lateral sclerosis mouse model. J. Neuroinflamm. 2014, 11, 42. [Google Scholar] [CrossRef]
- Almad, A.A.; Doreswamy, A.; Gross, S.K.; Richard, J.-P.; Huo, Y.; Haughey, N.; Maragakis, N.J. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 2016, 64, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastião, A.M.; Vaz, A.R.; Brites, D. Downregulated Glia Interplay and Increased miRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- D’hondt, C.; Ponsaerts, R.; De Smedt, H.; Bultynck, G.; Himpens, B. Pannexins, distant relatives of the connexin family with specific cellular functions? BioEssays 2009, 31, 953–974. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Cabré, R.; Alvarez, A.; Kong, M.; Burgos-Bravo, F.; Cárdenas, A.; Rojas-Mancilla, E.; Pérez-Nuñez, R.; Herrera-Molina, R.; Rojas, F.; Schneider, P.; et al. αVβ3 Integrin regulates astrocyte reactivity. J. Neuroinflam. 2017, 14, 194. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Chiot, A.; Lobsiger, C.S.; Boillée, S. New insights on the disease contribution of neuroinflammation in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2019, 32, 764–770. [Google Scholar] [CrossRef]
- Jones, M.K.; Nair, A.; Gupta, M. Mast Cells in Neurodegenerative Disease. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I. Amyotrophic Lateral Sclerosis, Neuroinflammation, and Cromolyn. Clin. Ther. 2020, 42, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Trias, E.; Kovacs, M.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Moura, I.C.; Hermine, O.; Beckman, J.S.; et al. Schwann cells orchestrate peripheral nerve inflammation through the expression of CSF1, IL-34, and SCF in amyotrophic lateral sclerosis. Glia 2020, 68, 1165–1181. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef]
- Staats, K.A.; Schönefeldt, S.; Van Helleputte, L.; Van Rillaer, M.; Lampi, Y.; Dooley, J.; Van Den Bosch, L.; Liston, A. C-kit is important for SOD1G93A mouse survival independent of mast cells. Neuroscience 2015, 301, 415–420. [Google Scholar] [CrossRef]
- Graves, M.; Fiala, M.; Dinglasan, L.A.; Liu, N.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2004, 5, 213–219. [Google Scholar] [CrossRef]
- Fiala, M.; Chattopadhay, M.; La Cava, A.; Tse, E.; Liu, G.; Lourenco, E.; Eskin, A.; Liu, P.T.; Magpantay, L.; Tse, S.; et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflam. 2010, 7, 76. [Google Scholar] [CrossRef]
- Brenner, T.; Soffer, D.; Shalit, M.; Levi-Schaffer, F. Mast cells in experimental allergic encephalomyelitis: Characterization, distribution in the CNS and in vitro activation by myelin basic protein and neuropeptides. J. Neurol. Sci. 1994, 122, 210–213. [Google Scholar] [CrossRef]
- Tanzola, M.B.; Robbie-Ryan, M.; Gutekunst, C.A.; Brown, M.A. Mast Cells Exert Effects Outside the Central Nervous System to Influence Experimental Allergic Encephalomyelitis Disease Course. J. Immunol. 2003, 171, 4385–4391. [Google Scholar] [CrossRef] [PubMed]
- Michaloudi, H.; Batzios, C.; Chiotelli, M.; Grivas, I.; Papadopoulos, G.C. Mast cells populations fluctuate along the spinal dura mater of the developing rat. Brain Res. 2008, 1226, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Granucci, E.J.; Griciuc, A.; Mueller, K.A.; Mills, A.N.; Le, H.; Dios, A.M.; McGinty, D.; Pereira, J.; Elmaleh, D.; Berry, J.D.; et al. Cromolyn sodium delays disease onset and is neuroprotective in the SOD1G93A Mouse Model of amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 17728. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harcha, P.A.; Garcés, P.; Arredondo, C.; Fernández, G.; Sáez, J.C.; van Zundert, B. Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions. Int. J. Mol. Sci. 2021, 22, 1924. https://doi.org/10.3390/ijms22041924
Harcha PA, Garcés P, Arredondo C, Fernández G, Sáez JC, van Zundert B. Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions. International Journal of Molecular Sciences. 2021; 22(4):1924. https://doi.org/10.3390/ijms22041924
Chicago/Turabian StyleHarcha, Paloma A., Polett Garcés, Cristian Arredondo, Germán Fernández, Juan C. Sáez, and Brigitte van Zundert. 2021. "Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions" International Journal of Molecular Sciences 22, no. 4: 1924. https://doi.org/10.3390/ijms22041924
APA StyleHarcha, P. A., Garcés, P., Arredondo, C., Fernández, G., Sáez, J. C., & van Zundert, B. (2021). Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions. International Journal of Molecular Sciences, 22(4), 1924. https://doi.org/10.3390/ijms22041924