
Comparing Super-Resolution Microscopy Techniques to Analyze Chromosomes

 , , and
, , and
Abstract
1. Introduction
2. Results
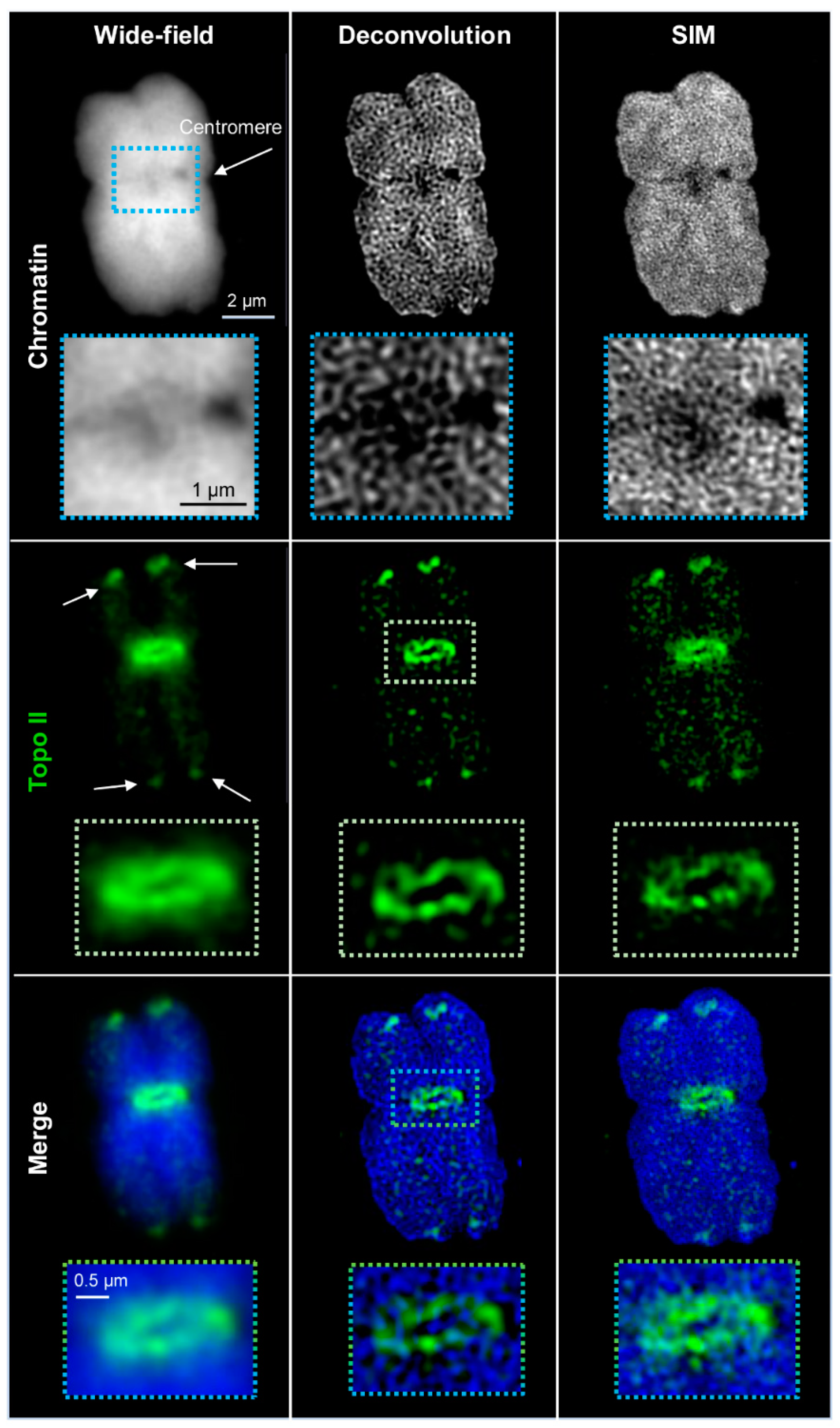
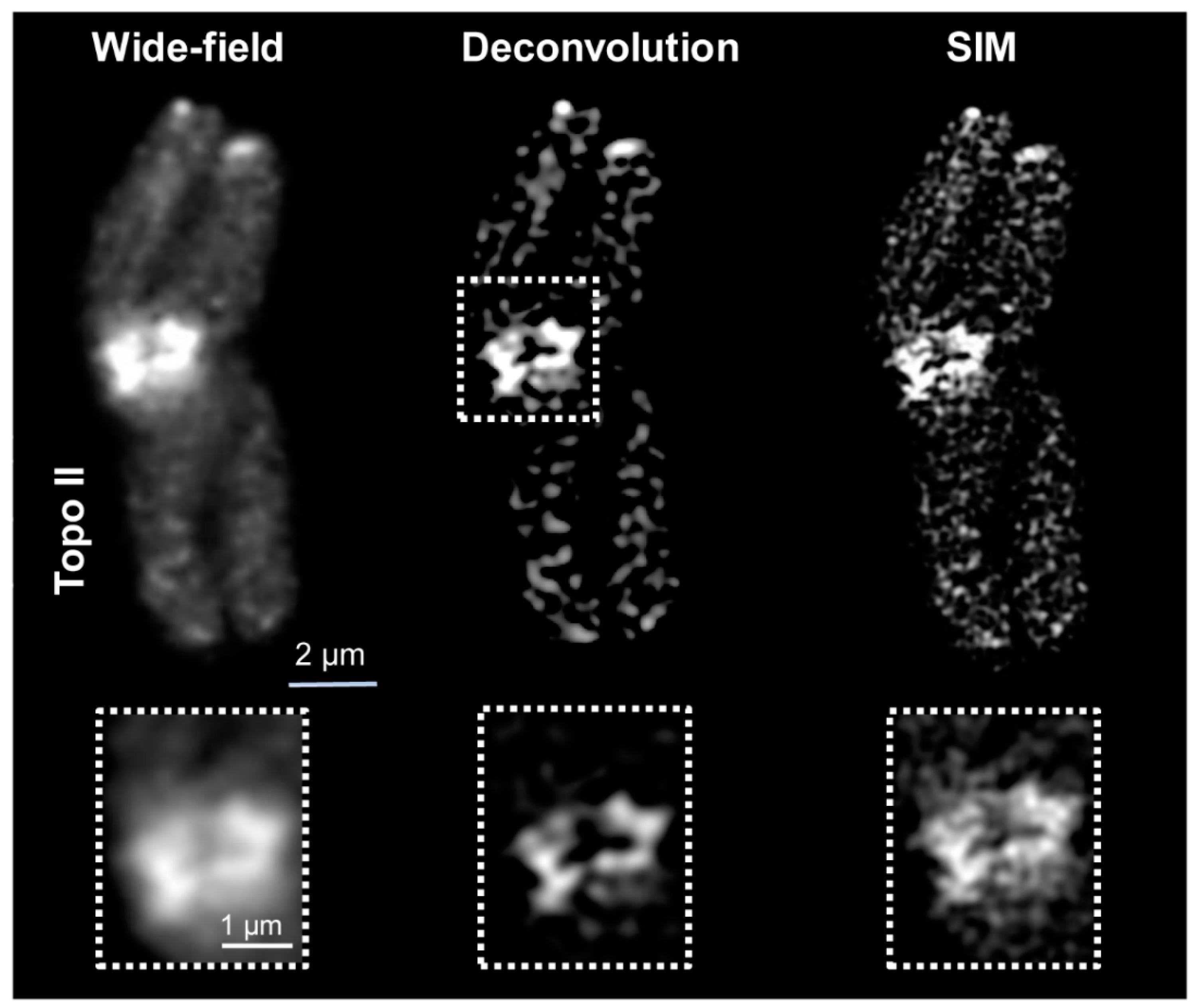
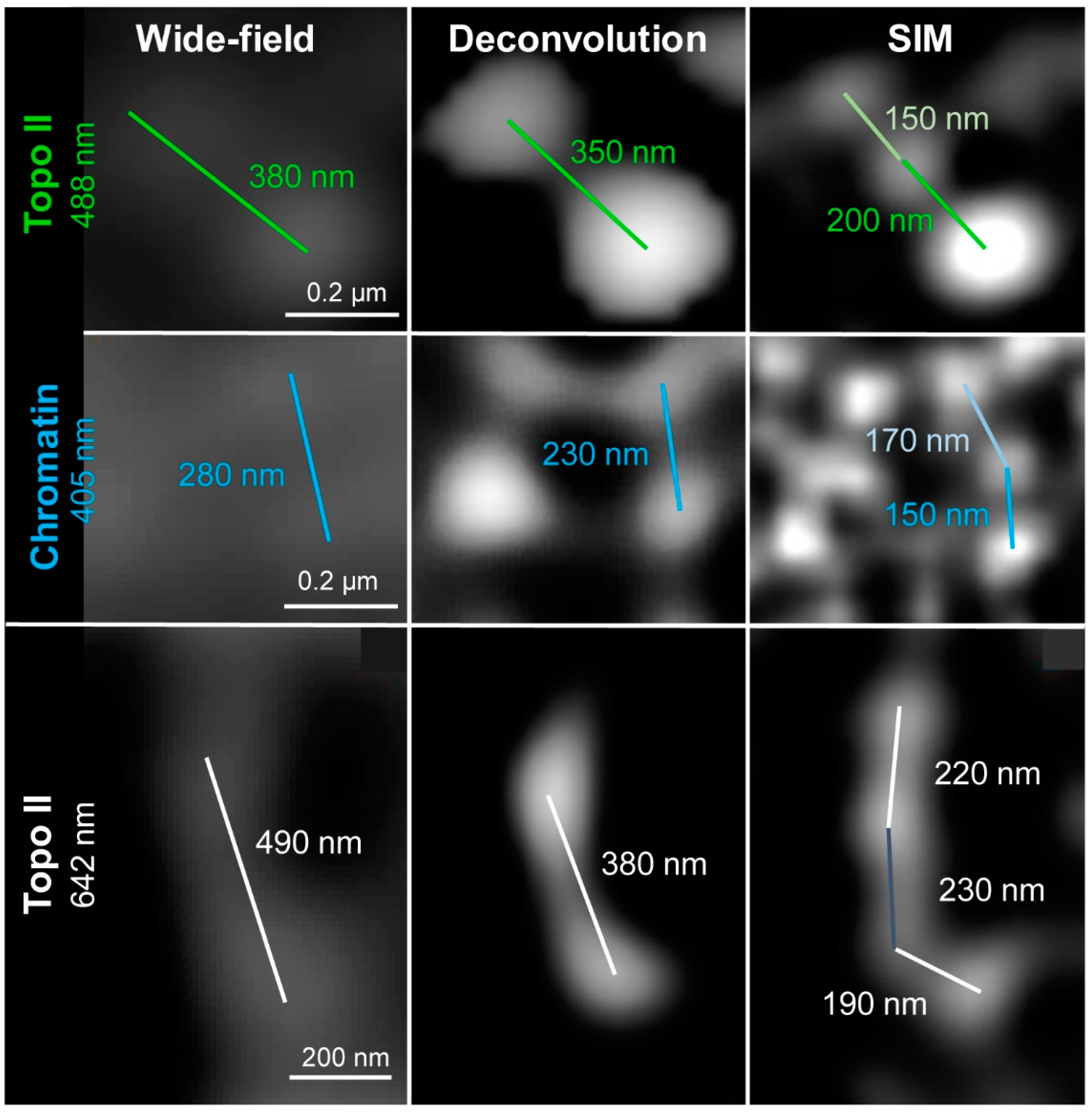
2.1. Wide-Field and SIM
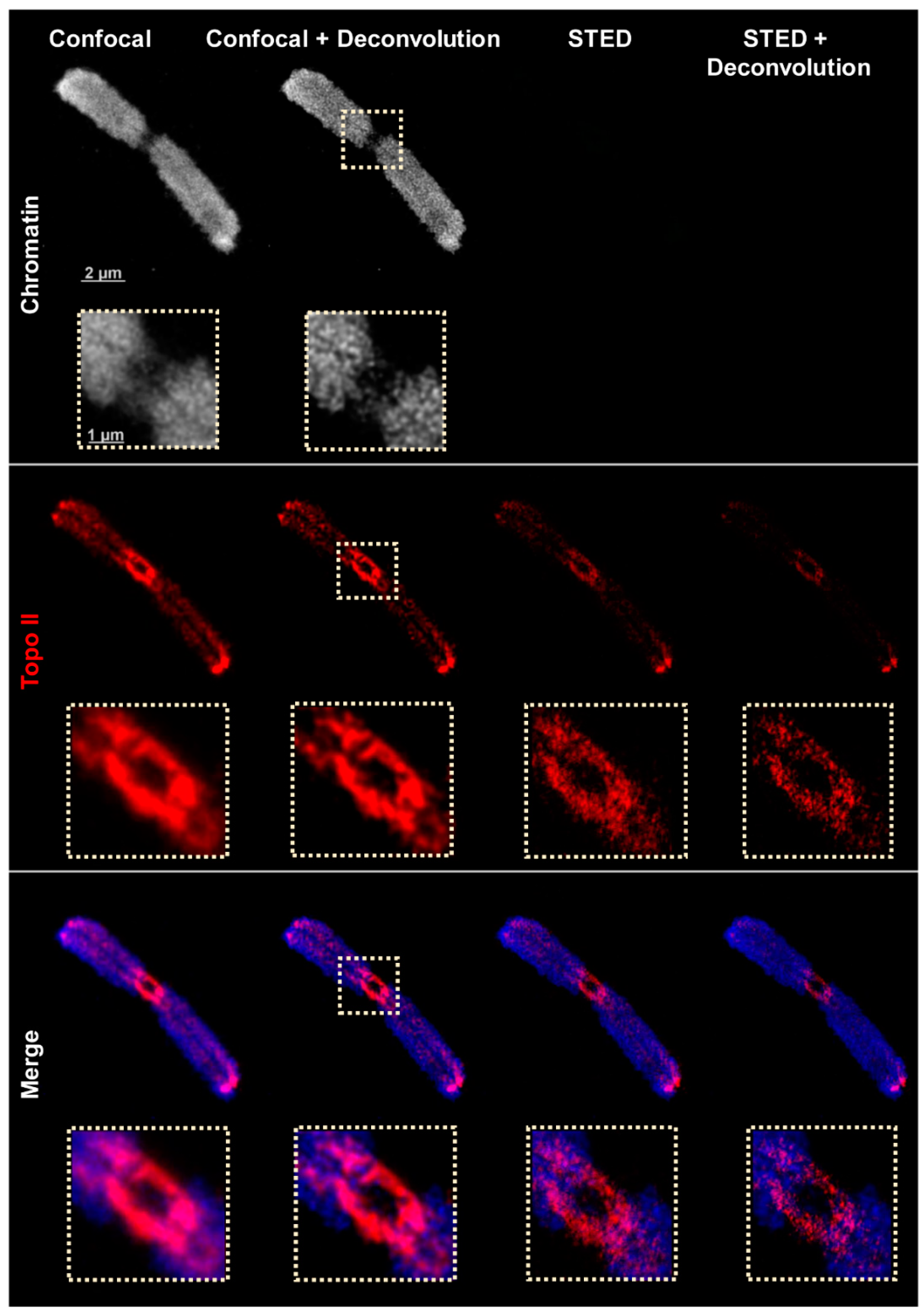
2.2. Confocal and STED Microscopy
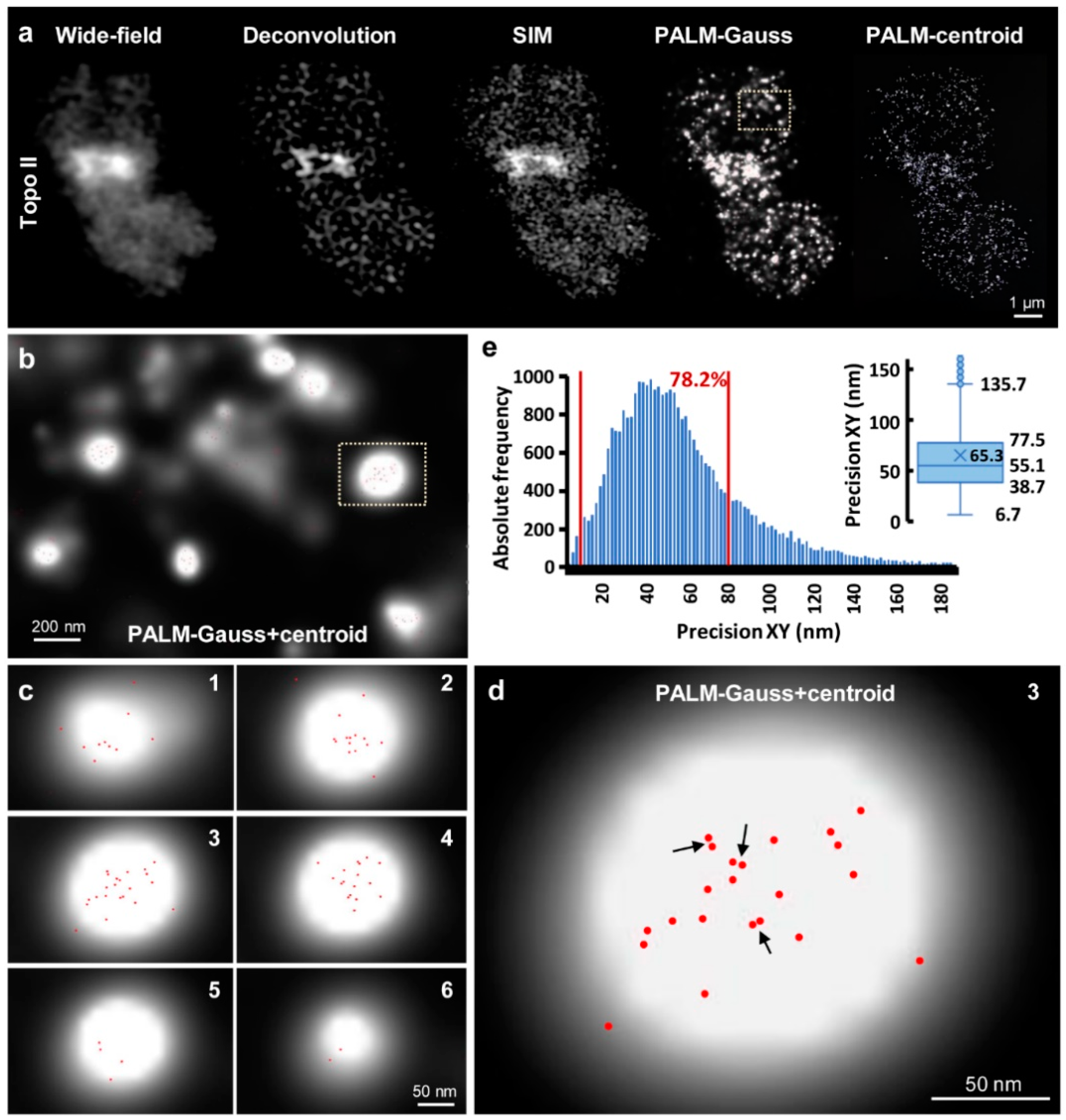
2.3. PALM
3. Discussion
4. Materials and Methods
4.1. Plant Material and Specimen Preparation
4.2. Topo II Antibody Preparation and Specificity Proof
4.3. Indirect Topo II Immunostaining for the Zeiss Elyra PS.1 Microscope System
4.4. Indirect Topo II Immunostaining for the Leica TCD SP8 STED 3X Microscope
4.5. Wide-Field, Deconvolution, 3D-SIM, and PALM
4.6. Confocal and STED Microscopy
4.7. Resolution Considerations
4.8. Resolution Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | three-dimensional |
| DCV | deconvolution |
| DAPI | 4′,6-diamidino-2-phenylindol |
| FWHM | full width at half maximum |
| gp | guinea pig |
| NOR | nucleolus organizer region |
| PALM | photoactivated localization microscopy |
| rb | rabbit |
| RT | room temperature |
| SIM | structured illumination microscopy |
| SMLM | single molecule localization microscopy |
| STED | stimulated emission depletion |
| STORM | stochastic optical reconstruction microscopy |
| Topo II | topoisomerase IIα |
References
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Archiv für Mikroskopische Anatomie 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Dodgson, J.; Chessel, A.; Cox, S.; Carazo Salas, R.E. Super-resolution microscopy: SIM, STED and Localization Microscopy. In Advanced Microscopy in Mycology; Dahms, T.E.S., Czymmek, K.J., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 47–60. [Google Scholar] [CrossRef]
- Pawley, J.B. Handbook of Biological Confocal Microscopy, 3rd ed.; Springer Science+Business Media LLC: New York, NY, USA, 2006. [Google Scholar] [CrossRef]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F.P. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 2018, 6, 022003. [Google Scholar] [CrossRef]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72–84. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Rego, E.H.; Shao, L.; Macklin, J.J.; Winoto, L.; Johansson, G.A.; Kamps-Hughes, N.; Davidson, M.W.; Gustafsson, M.G.L. Nonlinear structured-illumination microscopy with a photoswitchable protein reveals cellular structures at 50-nm resolution. Proc. Natl. Acad. Sci. USA 2011, 109, E135–E143. [Google Scholar] [CrossRef] [PubMed]
- Wegel, E.; Göhler, A.; Lagerholm, B.C.; Wainman, A.; Uphoff, S.; Kaufmann, R.; Dobbie, I.M. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Sci. Rep. 2016, 6, 27290. [Google Scholar] [CrossRef] [PubMed]
- Huff, J. The Airyscan detector: Confocal microscopy evolution for the neurosciences. Optoelectron. Gyroscopes 2018, 83–102. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the diffraction arrier: Super-resolution Imaging of Cells. Cell 2010, 143, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef]
- Liu, S.; Huh, H.; Lee, S.-H.; Huang, F. Three-dimensional single-molecule localization microscopy in whole-cell and tissue specimens. Annu. Rev. Biomed. Eng. 2020, 22, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Komis, G.; Šamajová, O.; Ovečka, M.; Šamaj, J. Super-resolution microscopy in plant cell imaging. Trends Plant Sci. 2015, 20, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Schubert, V. Super-resolution microscopy—Applications in plant cell research. Front. Plant Sci. 2017, 8, 531. [Google Scholar] [CrossRef]
- Korobchevskaya, K.; Lagerholm, B.C.; Colin-York, H.; Fritzsche, M. Exploring the potential of Airyscan microscopy for live cell imaging. Photonics 2017, 4, 41. [Google Scholar] [CrossRef]
- Müller, T.; Schumann, C.; Kraegeloh, A. STED Microscopy and its applications: New insights into cellular processes on the nanoscale. Chem. Phys. Chem. 2012, 13, 1986–2000. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Willig, K.I.; Harke, B.; Medda, R.; Hell, S.W. STED microscopy with continuous wave beams. Nat. Methods 2007, 4, 915–918. [Google Scholar] [CrossRef]
- Khater, I.M.; Nabi, I.R.; Hamarneh, G. A Review of super-resolution single-molecule localization microscopy cluster analysis and quantification methods. Gene Exp. Patterns 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xin, B.; Kuang, W.; Zhou, Z.; Huang, Z.-L. Divide and conquer: Real-time maximum likelihood fitting of multiple emitters for super-resolution localization microscopy. Opt. Express 2019, 27, 21029–21049. [Google Scholar] [CrossRef]
- Weisshart, K.; Dertinger, T.; Kalkbrenner, T.; Kleppe, I.; Kempe, M. Super-resolution microscopy heads towards 3D dynamics. Adv. Opt. Technol. 2013, 2, 211–231. [Google Scholar] [CrossRef]
- Komis, G.; Novák, D.; Ovečka, M.; Šamajová, O.; Šamaj, J. Advances in imaging plant cell dynamics. Plant Physiol. 2018, 176, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Kostiuk, G.; Bucevicius, J.; Gerasimaite, R.; Lukinavicius, G. Application of STED imaging for chromatin studies. J. Phys. D Appl. Phys. 2019, 52, 504003. [Google Scholar] [CrossRef]
- Maß, L.; Holtmannspötter, M.; Zachgo, S. Dual-color 3D-dSTORM colocalization and quantification of ROXY1 and RNAPII variants throughout the transcription cycle in root meristem nuclei. Plant J. 2020, 104, 1423–1436. [Google Scholar] [CrossRef]
- Němečková, A.; Wäsch, C.; Schubert, V.; Ishii, T.; Hřibová, E.; Houben, A. CRISPR/Cas9-based RGEN-ISL allows the simultaneous and specific visualization of proteins, DNA repeats, and sites of DNA replication. Cytogenet. Genome Res. 2019, 159, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Schubert, V.; Neumann, P.; Marques, A.; Heckmann, S.; Macas, J.; Pedrosa-Harand, A.; Schubert, I.; Jang, T.-S.; Houben, A. Super-resolution microscopy reveals diversity of plant centromere architecture. Int. J. Mol. Sci. 2020, 21, 3488. [Google Scholar] [CrossRef] [PubMed]
- Schubert, V.; Weisshart, K. Abundance and distribution of RNA polymerase II in Arabidopsis interphase nuclei. J. Exp. Bot. 2015, 66, 1687–1698. [Google Scholar] [CrossRef]
- Tichá, M.; Hlaváčková, K.; Hrbáčková, M.; Ovečka, M.; Šamajová, O.; Šamaj, J. Super-resolution imaging of microtubules in Medicago Sativa. Methods Cell Biol. 2020, 160, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Sivaguru, M.; Urban, M.A.; Fried, G.; Wesseln, C.J.; Mander, L.; Punyasena, S.W. Comparative performance of airyscan and structured illumination superresolution microscopy in the study of the surface texture and 3D shape of pollen. Microsc. Res. Tech. 2018, 81, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Tröger, J.; Hoischen, C.; Perner, B.; Monajembashi, S.; Barbotin, A.; Löschberger, A.; Eggeling, C.; Kessels, M.M.; Qualmann, B.; Hemmerich, P. Comparison of mMultiscale imaging methods for brain research. Cells 2020, 9, 1377. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Uchiyama, S. Chromosome protein framework from proteome analysis of isolated human metaphase chromosomes. Chem. Rec. 2007, 7, 230–237. [Google Scholar] [CrossRef]
- Hizume, K.; Araki, S.; Yoshikawa, K.; Takeyasu, K. Topoisomerase II, scaffold component, promotes chromatin compaction in vitro in a linker-histone H1-dependent manner. Nucleic Acids Res. 2007, 35, 2787–2799. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Liang, Z.; Mukhina, M.; Fisher, J.; Vincenten, N.; Zhang, Z.; Hutchinson, J.; Zickler, D.; Kleckner, N. The 3D topography of Mmitotic chromosomes. Mol. Cell 2020, 79, 902–916.e6. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Halligan, B.; Cooke, C.A.; Heck, M.M.S.; Liu, L.F. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell Biol. 1985, 100, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Samejima, K.; Samejima, I.; Vagnarelli, P.; Ogawa, H.; Vargiu, G.; Kelly, D.A.; Alves, F.D.L.; Kerr, A.; Green, L.C.; Hudson, D.F.; et al. Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIα. J. Cell Biol. 2012, 199, 755–770. [Google Scholar] [CrossRef]
- Żabka, A.; Polit, J.T.; Bernasińska, J.; Maszewski, J. DNA topoisomerase II-dependent control of the cell cycle progression in root meristems of Allium cepa. Cell Biol. Int. 2013, 38, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Achary, V.M.M.; Panditi, V.; Sopory, S.K.; Reddy, M.K. Dynamics of tobacco DNA topoisomerases II in cell cycle regulation: To manage topological constrains during replication, transcription and mitotic chromosome condensation and segregation. Plant Mol. Biol. 2017, 94, 595–607. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.; Schubert, V.; Osman, K.; Darbyshire, A.; Sanchez-Moran, E.; Franklin, F.C.H. TOPII and chromosome movement help remove interlocks between entangled chromosomes during meiosis. J. Cell Biol. 2018, 217, 4070–4079. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, C.M. On spectroscopic resolving power. Astrophys. J. 1916, 44, 76. [Google Scholar] [CrossRef]
- Schultz, P.; Olland, S.; Oudet, P.; Hancock, R. Structure and conformational changes of DNA topoisomerase II visualized by electron microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 5936–5940. [Google Scholar] [CrossRef]
- Hebisch, E.; Wagner, E.; Westphal, V.; Sieber, J.; Lehnart, S. A protocol for registration and correction of multicolour STED superresolution images. J. Microsc. 2017, 267, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Weisshart, K.; Fuchs, J.; Schubert, V. Structured illumination microscopy (SIM) and photoactivated localization microscopy (PALM) to analyze the abundance and distribution of RNA polymerase II molecules on flow-sorted Arabidopsis nuclei. Bio-Protocol 2016, 6, 1725. [Google Scholar] [CrossRef]
- Lysák, M.A.; Číhalíková, J.; Kubaláková, M.; Šimková, H.; Künzel, G.; Doležel, J. Flow karyotyping and sorting of mitotic chromosomes of barley (Hordeum vulgare L.). Chromosome Res. 1999, 7, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Karras, C.; Smedh, M.; Förster, R.; Deschout, H.; Fernandez-Rodriguez, J.; Heintzmann, R. Successful optimization of reconstruction parameters in structured illumination microscopy—A practical guide. Opt. Commun. 2019, 436, 69–75. [Google Scholar] [CrossRef]
- Dekker, A.J.D.; Bos, A.V.D. Resolution: A survey. J. Opt. Soc. Am. A 1997, 14, 547–557. [Google Scholar] [CrossRef]
- Weik, M.H. Nyquist theorem. In Computer Science and Communications Dictionary; Springer International Publishing: Boston, MA, USA, 2000; p. 1127. [Google Scholar]
- Nieuwenhuizen, R.P.; Stallinga, S.; Rieger, B. Visualization and resolution in localization microscopy. In Cell Membrane Nanodomains; CRC Press: New York, NY, USA, 2015. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microscopy | Lateral Resolution | Axial Resolution | Platform | Fluorochrome Requirement |
|---|---|---|---|---|
| WF | ~200–300 | ~500–700 | Wide-field | Standard |
| CLSM | ~150–220 | ~360–500 | Confocal | Standard |
| SIM | ~100–120 | ~350–400 | Wide-field | Standard |
| STED | ~70–90 * | 100–200 ** | Confocal | Photostable |
| PALM | ~20–50 | ~10–70 | Wide-field | Switchable |
| Achieved Resolution | DAPI (nm) λem = 461 nm | Alexa488 (nm) λem = 520 nm | STAR635P (nm) λem = 651 nm |
|---|---|---|---|
| Wide-field | 297 ± 11 (6) | 421 ± 18 (6) | 499 ± 22 (7) |
| Deconvolution | 216 ± 6 (6) | 303 ± 11 (6) | 363 ± 9 (7) |
| SIM | 131 ± 9 (6) | 171 ± 7 (6) | 234 ± 21 (7) |
| Theoretical Resolution w/NA 1.4 | |||
| WF (Sparrow) | ~160 | ~180 | ~220 |
| DCV (WF/√2) | ~110 | ~130 | ~155 |
| SIM (WF/2) | ~80 | ~90 | ~110 |
| WF (Raleigh) | ~200 | ~230 | ~290 |
| DCV (WF/√2) | ~140 | ~165 | ~205 |
| SIM (WF/2) | ~100 | ~115 | ~145 |
| Resolution | DABCO (nm) | Diamond (nm) |
|---|---|---|
| Confocal | 468 ± 38 (6) | 432 ± 36 (6) |
| Deconvolution | 243 ± 22 (6) | 237 ± 23 (6) |
| STED | 138 ± 23 (6) | 134 ± 17 (6) |
| STED + Deconvolution | 78 ± 15 (6) | 72 ± 12 (6) |
| Criterium | Lateral Resolution | Axial Resolution |
|---|---|---|
| Raleigh | ||
| FWHM | ||
| Sparrow |
| Method | Improvement Factor | Parameter |
|---|---|---|
| DCV | Fixed number | |
| CLSM | Fixed number | |
| STED | I = intracavity intensity Isat = saturation intensity g = geometric factor | |
| SIM | Fixed number | |
| SMLM | N = number of photons g = geometric factor |
| Beam Width | Definition | % of Maximum |
|---|---|---|
| Sparrow | 98.6 | |
| SD | 60.7 | |
| FWHM | 50 | |
| Raleigh | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubalová, I.; Němečková, A.; Weisshart, K.; Hřibová, E.; Schubert, V. Comparing Super-Resolution Microscopy Techniques to Analyze Chromosomes. Int. J. Mol. Sci. 2021, 22, 1903. https://doi.org/10.3390/ijms22041903
Kubalová I, Němečková A, Weisshart K, Hřibová E, Schubert V. Comparing Super-Resolution Microscopy Techniques to Analyze Chromosomes. International Journal of Molecular Sciences. 2021; 22(4):1903. https://doi.org/10.3390/ijms22041903
Chicago/Turabian StyleKubalová, Ivona, Alžběta Němečková, Klaus Weisshart, Eva Hřibová, and Veit Schubert. 2021. "Comparing Super-Resolution Microscopy Techniques to Analyze Chromosomes" International Journal of Molecular Sciences 22, no. 4: 1903. https://doi.org/10.3390/ijms22041903
APA StyleKubalová, I., Němečková, A., Weisshart, K., Hřibová, E., & Schubert, V. (2021). Comparing Super-Resolution Microscopy Techniques to Analyze Chromosomes. International Journal of Molecular Sciences, 22(4), 1903. https://doi.org/10.3390/ijms22041903