
Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes


Abstract
1. Introduction
2. Results
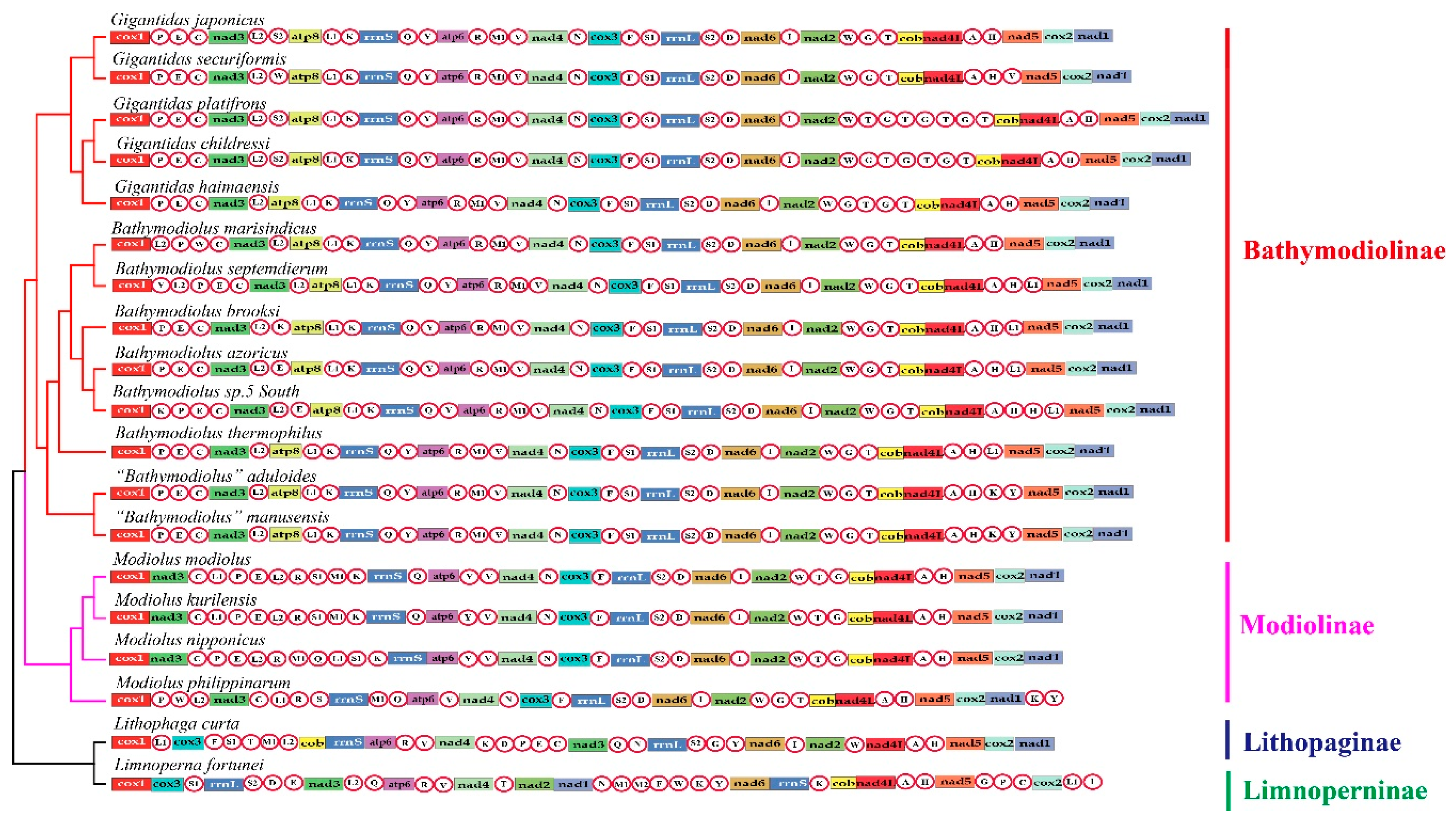
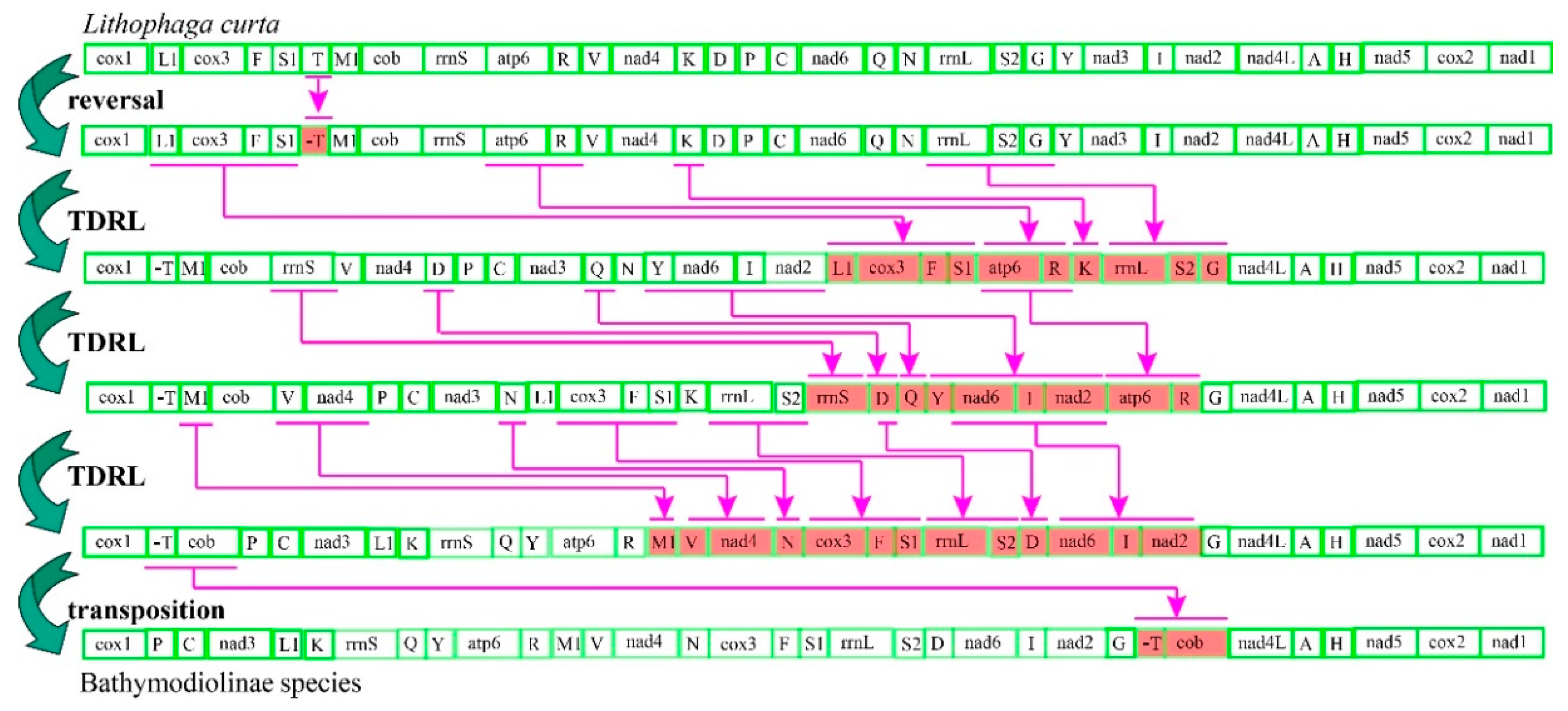
2.1. Genome Organizations and Gene Rearrangement
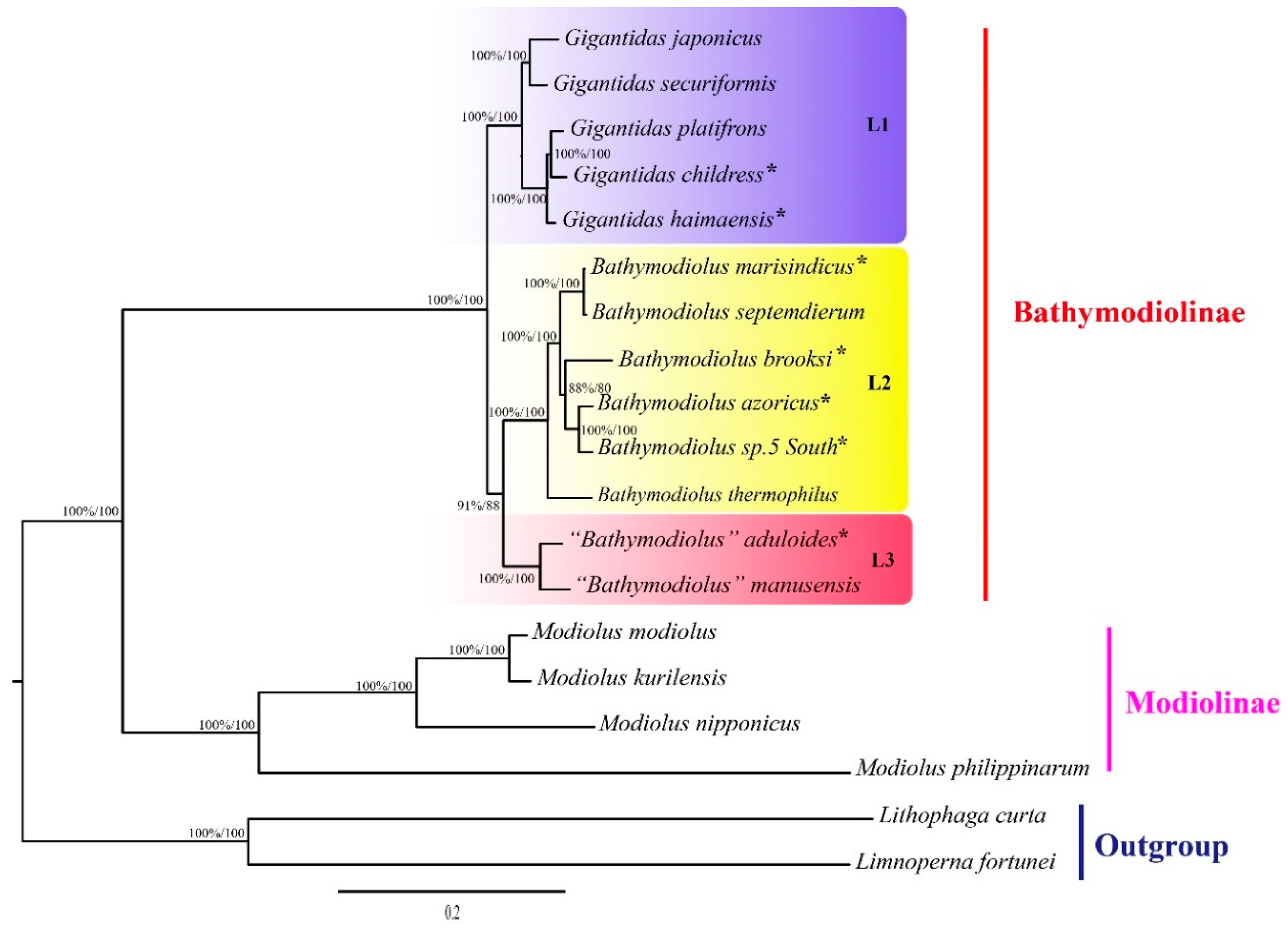
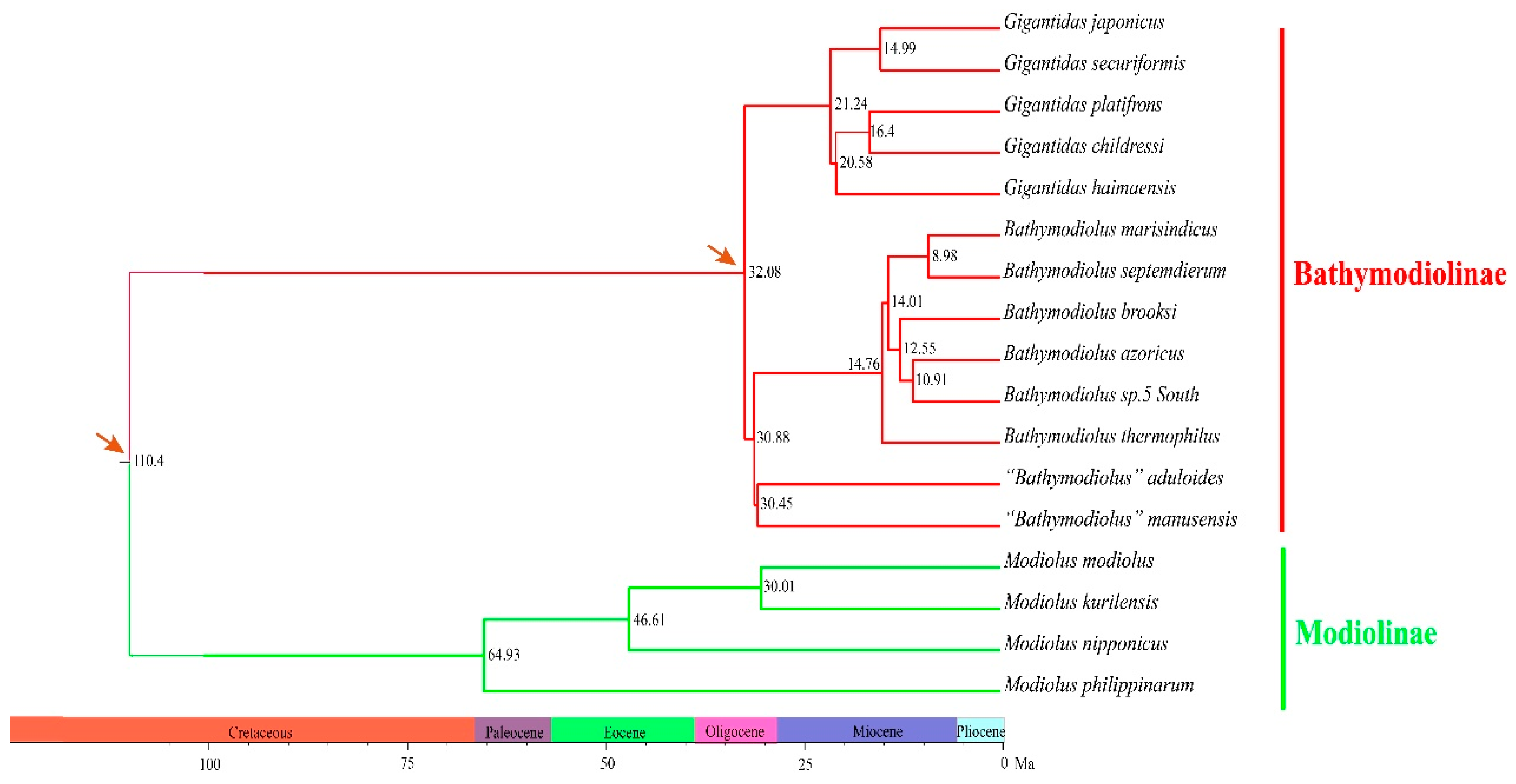
2.2. Phylogenetic Relationships and Divergence Times
2.3. Genetic Distance
2.4. Elevated Nucleotide Substitution Rates in Deep-Sea Mussels
3. Discussion
3.1. General Features of Bathymodiolinae Mitogenomes
3.2. Molecular Phylogeny of Deep-Sea Mussels
3.3. Mitochondrial Gene Rearrangement
3.4. Adaptations to Deep-Sea Environments
4. Materials and Methods
4.1. Acquisition of Mitochondrial Genome Sequences
4.2. Genome Sequence Annotation and Gene Arrangement Analysis
4.3. Phylogenetic Analyses and Divergence Time Estimation
4.4. Positive Selection Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Distel, D.L.; Baco, A.R.; Chuang, E.; Morrill, W.; Cavanaugh, C.; Smith, C.R. Marine ecology: Do mussels take wooden steps to deep-sea vents? Nature 2000, 403, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kwak, H.; Shin, J.; Kim, S.C.; Kim, T.; Park, J.K. A mitochondrial genome phylogeny of Mytilidae (Bivalvia: Mytilida). Mol. Phylogenet. Evol. 2019, 139, 106533. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, H.; Zhang, H. Phylogeny and evolutionary radiation of the marine mussels (Bivalvia: Mytilidae) based on mitochondrial and nuclear genes. Mol. Phylogenet. Evol. 2018, 126, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Lorion, J.; Kiel, S.; Faure, B.; Kawato, M.; Ho, S.Y.; Marshall, B.; Tsuchida, S.; Miyazaki, J.I.; Fujiwara, Y. Adaptive radiation of chemosymbiotic deep-sea mussels. Proc. R. Soc. B-Biol. Sci. 2013, 280, 20131243. [Google Scholar] [CrossRef] [PubMed]
- Thubaut, J.; Puillandre, N.; Faure, B.; Cruaud, C.; Samadi, S. The contrasted evolutionary fates of deep-sea chemosynthetic mussels (Bivalvia, Bathymodiolinae). Ecol. Evol. 2013, 3, 4748–4766. [Google Scholar] [CrossRef]
- Lorion, J.; Buge, B.; Cruaud, C.; Samadi, S. New insights into diversity and evolution of deep-sea Mytilidae (Mollusca: Bivalvia). Mol. Phylogenet. Evol. 2010, 57, 71–83. [Google Scholar] [CrossRef]
- Johnson, S.; Won, Y.-J.; Harvey, J.; Vrijenhoek, R. A hybrid zone between Bathymodiolus mussel lineages from eastern Pacific hydrothermal vents. BMC Evol. Biol. 2013, 13, 21. [Google Scholar] [CrossRef]
- Bougerol, M.; Boutet, I.; LeGuen, D.; Jollivet, D.; Tanguy, A. Transcriptomic response of the hydrothermal mussel Bathymodiolus azoricus in experimental exposure to heavy metals is modulated by the Pgm genotype and symbiont content. Mar. Genomics 2015, 21, 63–73. [Google Scholar] [CrossRef]
- Bettencourt, R.; Pinheiro, M.; Egas, C.; Gomes, P.; Afonso, M.; Shank, T.; Santos, R. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics 2010, 11, 559. [Google Scholar] [CrossRef]
- Genio, L.; Kiel, S.; Cunha, M.R.; Grahame, J.; Little, C.T. Shell microstructures of mussels (Bivalvia: Mytilidae: Bathymodiolinae) from deep-sea chemosynthetic sites: Do they have a phylogenetic significance? Deep Sea Res. Part I 2012, 64, 86–103. [Google Scholar] [CrossRef]
- Xu, T.; Feng, D.; Tao, J.; Qiu, J.W. A new species of deep-sea mussel (Bivalvia: Mytilidae: Gigantidas) from the South China Sea: Morphology, phylogenetic position, and gill-associated microbes. Deep Sea Res. Part I 2019, 146, 79–90. [Google Scholar] [CrossRef]
- Samadi, S.; Puillandre, N.; Pante, E.; Boisselier, M.C.; Corbari, L.; Chen, W.J.; Maestrati, P.; Mana, R.; Thubaut, J.; Zuccon, D.; et al. Patchiness of deep-sea communities in Papua New Guinea and potential susceptibility to anthropogenic disturbances illustrated by seep organisms. Mar. Ecol. 2015, 36, 109–132. [Google Scholar] [CrossRef]
- Gvoždík, V.; Moravec, J.; Klütsch, C.; Kotlík, P. Phylogeography of the Middle Eastern tree frogs (Hyla, Hylidae, Amphibia) as inferred from nuclear and mitochondrial DNA variation, with a description of a new species. Mol. Phylogenet. Evol. 2010, 55, 1146–1166. [Google Scholar] [CrossRef]
- Lei, R.; Shore, G.D.; Brenneman, R.A.; Engberg, S.E.; Sitzmann, B.D.; Bailey, C.A.; Kimmel, L.M.; Randriamampionona, R.; Ranaivoarisoa, J.F.; Louis, E.E., Jr. Complete sequence and gene organization of the mitochondrial genome for Hubbard’s sportive lemur (Lepilemur hubbardorum). Gene 2010, 464, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.P.; Xin, Z.Z.; Liu, Y.; Zhang, D.Z.; Wang, Z.F.; Zhang, H.B.; Chai, X.Y.; Zhou, C.L.; Liu, Q.N. The complete mitochondrial genome of Sesarmops sinensis reveals gene rearrangements and phylogenetic relationships in Brachyura. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Li, J.Y.; Song, Z.L.; Yan, G.Y.; He, L.S. The complete mitochondrial genome of the largest amphipod, Alicella gigantea: Insight into its phylogenetic relationships and deep-sea adaptive characters. Int. J. Biol. Macromol. 2019, 141, 570–577. [Google Scholar] [CrossRef]
- Mu, W.; Liu, J.; Zhang, H. Complete mitochondrial genome of Benthodytes marianensis (Holothuroidea: Elasipodida: Psychropotidae): Insight into deep sea adaptation in the sea cucumber. PLoS ONE 2018, 13, e0208051. [Google Scholar] [CrossRef]
- Shen, X.; Pu, Z.; Chen, X.; Murphy, R.W.; Shen, Y. Convergent Evolution of Mitochondrial Genes in Deep-Sea Fishes. Front. Genet 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, Y.; Wang, X.; Jiang, W.; Yin, J.; Lin, Q. Comparative analysis of mitochondrial genome of a deep-sea crab Chaceon granulates reveals positive selection and novel genetic features. J. Oceanol. Limnol. 2019, 38, 427–437. [Google Scholar] [CrossRef]
- Yang, M.; Gong, L.; Sui, J.; Li, X. The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS ONE 2019, 14, e0217952. [Google Scholar] [CrossRef]
- Boore, J.L.; Medina, M.; Rosenberg, L.A. Complete sequences of the highly rearranged molluscan mitochondrial genomes of the scaphopod Graptacme eborea and the bivalve Mytilus edulis. Mol. Biol. Evol. 2004, 21, 1492–1503. [Google Scholar] [CrossRef]
- Breton, S.; Stewart, D.T.; Hoeh, W.R. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of Mytilus spp. (Bivalvia: Mytilidae): Identification of the “missing” ATPase 8 gene. Mar. Genomics 2010, 3, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, G.; Shimamura, S.; Takaki, Y.; Yokobori, S.I.; Ohara, Y.; Takishita, K.; Maruyama, T.; Fujikura, K.; Yoshida, T. Updated mitochondrial phylogeny of Pteriomorph and Heterodont Bivalvia, including deep-sea chemosymbiotic Bathymodiolus mussels, vesicomyid clams and the thyasirid clam Conchocele cf. bisecta. Mar. Genomics 2017, 31, 43–52. [Google Scholar] [CrossRef]
- Uliano-Silva, M.; Americo, J.A.; Costa, I.; Schomaker-Bastos, A.; de Freitas Rebelo, M.; Prosdocimi, F. The complete mitochondrial genome of the golden mussel Limnoperna fortunei and comparative mitogenomics of Mytilidae. Gene 2016, 577, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, J.I.; de Oliveira Martins, L.; Fujita, Y.; Matsumoto, H.; Fujiwara, Y. Evolutionary process of deep-sea Bathymodiolus mussels. PLoS ONE 2010, 5, e10363. [Google Scholar] [CrossRef]
- Savolainen, V.; Cowan, R.S.; Vogler, A.P.; Roderick, G.K.; Lane, R. Towards writing the encyclopaedia of life: An introduction to DNA barcoding. Philos. Trans. R. Soc. B 2005, 360, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhou, Y.; Chen, C.; Kwan, Y.H.; Sun, Y.; Wang, X.; Yang, L.; Zhang, R.; Wei, T.; Yang, Y.; et al. Nearest vent, dearest friend: Biodiversity of Tiancheng vent field reveals cross-ridge similarities in the Indian Ocean. R. Soc. Open. Sci. 2020, 7, 200110. [Google Scholar] [PubMed]
- Akasaki, T.; Nikaido, M.; Tsuchiya, K.; Segawa, S.; Hasegawa, M.; Okada, N. Extensive mitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications. Mol. Phylogenet. Evol. 2006, 38, 648–658. [Google Scholar] [CrossRef]
- Ki, J.S.; Lee, Y.M.; Jung, S.O.; Horiguchi, T.; Cho, H.S.; Lee, J.S. Mitochondrial genome of Thais clavigera (Mollusca: Gastropoda): Affirmation of the conserved, ancestral gene pattern within the mollusks. Mol. Phylogenet. Evol. 2010, 54, 1016–1020. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Xie, G.L.; Köhler, F.; Huang, X.C.; Wu, R.W.; Zhou, C.H.; Ouyang, S.; Wu, X.P. A novel gene arrangement among the Stylommatophora by the complete mitochondrial genome of the terrestrial slug Meghimatium bilineatum (Gastropoda, Arionoidea). Mol. Phylogenet. Evol. 2019, 135, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Zeng, C.; Yan, G.Y.; He, L.S. Characterization of the mitochondrial genome of an ancient amphipod Halice sp. MT-2017 (Pardaliscidae) from 10,908 m in the Mariana Trench. Sci. Rep. UK 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hogan, R.I.; Hopkins, K.; Wheeler, A.J.; Allcock, A.L.; Yesson, C. Novel diversity in mitochondrial genomes of deep-sea Pennatulacea (Cnidaria: Anthozoa: Octocorallia). Mitochondrial DNA A 2019, 30, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, J.; Rouse, G.W.; Wiklund, H.; Pleijel, F.; Watanabe, H.K.; Chen, C.; Qian, P.Y.; Qiu, J.W. Phylogeny, evolution and mitochondrial gene order rearrangement in scale worms (Aphroditiformia, Annelida). Mol. Phylogenet. Evol. 2019, 125, 220–231. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Plazzi, F.; Puccio, G.; Passamonti, M. Burrowers from the past: Mitochondrial signatures of Ordovician bivalve infaunalization. Genome Biol. Evol. 2017, 9, 956–967. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, X.; Sun, L.; Bai, Y.; Zhang, D.; Tang, B. Evolution of mitochondrial energy metabolism genes associated with hydrothermal vent adaption of Alvinocaridid shrimps. Genes Genomics 2017, 39, 1367–1376. [Google Scholar] [CrossRef]
- Hui, M.; Wang, M.; Sha, Z. The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comp. Biochem. Physiol. Part D Genomics Proteomics 2018, 25, 42–52. [Google Scholar]
- Shen, Y.; Kou, Q.; Zhong, Z.; Li, X.; He, L.; He, S.; Gan, X. The first complete mitogenome of the South China deep-sea giant isopod Bathynomus sp. (Crustacea: Isopoda: Cirolanidae) allows insights into the early mitogenomic evolution of isopods. Ecol. Evol. 2017, 7, 1869–1881. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Liang, L.; Zhu, Z.H.; Zhou, W.P.; Irwin, D.M.; Zhang, Y.P. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 2010, 107, 8666–8671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Ning, T.; Xiao, H.; Li, J.; Hua, S.; Zhang, Y.P. Adaptive evolution of the mitochondrial ND6 gene in the domestic horse. Genet. Mol. Res. 2010, 9, 144–150. [Google Scholar] [CrossRef]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, W.; Gao, Y.; Tang, S.; Huang, Q.; Tan, X.; Chen, J.; Huang, T. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion 2008, 8, 352–357. [Google Scholar] [CrossRef]
- Mahalingam, S.; McClelland, G.B.; Scott, G.R. Evolved changes in the intracellular distribution and physiology of muscle mitochondria in high-altitude native deer mice. J. Physiol. 2017, 595, 4785–4801. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Li, Y.; Ruan, Z.; Bian, C.; You, X.; Yang, J.; Jiang, W.; Shi, Q. The Complete Mitochondrial Genome of Glyptothorax macromaculatus Provides a Well-Resolved Molecular Phylogeny of the Chinese Sisorid Catfishes. Genes 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, F.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. GBLOCKS: Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Version 0.91 b; EMBL: Heidelberg, Germany, 2002. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 46, W537–W545. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [PubMed]
- Rambaut, A. FigTree v1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 4 June 2020).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Subfamily | bp | Accession No. | Reference |
|---|---|---|---|---|
| Gigantidas japonicus | Bathymodiolinae | 17,510 | AP014560 | Robicheau et al. (2017) |
| Gigantidas platifrons | Bathymodiolinae | 17,653 | AP014561 | Robicheau et al. (2017) |
| Bathmodiolus septemdierm | Bathymodiolinae | 17,069 | AP014562 | Robicheau et al. (2017) |
| Bathymodiolus thermophilus | Bathymodiolinae | 18,819 | MK721544 | Lee et al. (2019) |
| Bathymodiolus securiformis | Bathymodiolinae | 17,199 | NC_039552 | - |
| Bathymodiolus manusensis | Bathymodiolinae | 16,801 | KY270856 | - |
| Bathymodiolus sp. 5 South | Bathymodiolinae | 18,376 | MT916740 | This study |
| Bathymodiolus aduloides | Bathymodiolinae | 17,243 | MT916741 | This study |
| Bathymodiolus azoricus | Bathymodiolinae | 17,598 | MT916742 | This study |
| Bathymodiolus brooksi | Bathymodiolinae | 17,728 | MT916743 | This study |
| Bathymodiolus childressi | Bathymodiolinae | 17,637 | MT916744 | This study |
| Bathymodiolus marisindicus | Bathymodiolinae | 17,138 | MT916745 | This study |
| Gigantidas haimaensis | Bathymodiolinae | 18,283 | MT916746 | This study |
| Modiolus modiolus | Modiolinae | 15,816 | KX821782 | Robicheau et al. (2017) |
| Modiolus kurilensis | Modiolinae | 16,210 | KY242717 | - |
| Modiolus nipponicus | Modiolinae | 15,638 | MK721547 | Lee et al. (2019) |
| Modiolus philippinarum | Modiolinae | 16,389 | KY705073 | Sun et al. (2017) |
| Lithophaga curta | Lithophaginae | 16,580 | MK721546 | Lee et al. (2019) |
| Limnoperna fortunei | Limnoperninae | 18,145 | KP756905 | Uliano-Silva et al. (2016) |
| Gene | Codon | Amino Acid | BEB Values |
|---|---|---|---|
| atp6 | 50 | L | 0.994 |
| nad4 | 76 | P | 0.992 |
| 234 | K | 0.991 | |
| nad2 | 34 | S | 0.991 |
| 37 | S | 0.995 | |
| 59 | K | 0.991 | |
| 60 | S | 0.994 | |
| 122 | W | 0.997 | |
| 331 | G | 0.992 | |
| cob | 54 | S | 0.991 |
| 317 | N | 0.991 | |
| 351 | L | 0.994 | |
| nad5 | 85 | S | 0.995 |
| 439 | S | 0.997 | |
| 503 | S | 0.991 | |
| cox2 | 95 | K | 0.993 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Sun, J.; Xu, T.; Qiu, J.-W.; Qian, P.-Y. Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes. Int. J. Mol. Sci. 2021, 22, 1900. https://doi.org/10.3390/ijms22041900
Zhang K, Sun J, Xu T, Qiu J-W, Qian P-Y. Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes. International Journal of Molecular Sciences. 2021; 22(4):1900. https://doi.org/10.3390/ijms22041900
Chicago/Turabian StyleZhang, Kai, Jin Sun, Ting Xu, Jian-Wen Qiu, and Pei-Yuan Qian. 2021. "Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes" International Journal of Molecular Sciences 22, no. 4: 1900. https://doi.org/10.3390/ijms22041900
APA StyleZhang, K., Sun, J., Xu, T., Qiu, J.-W., & Qian, P.-Y. (2021). Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes. International Journal of Molecular Sciences, 22(4), 1900. https://doi.org/10.3390/ijms22041900