
Role of the Macrophage Migration Inhibitory Factor in the Pathophysiology of Pre-Eclampsia

 ,
, 
 , , ,
, , ,
Abstract
1. Introduction
2. Immune Response in Pregnancy
2.1. Normal Pregnancy
2.2. Pre-Eclampsia
3. MIF in Normal and Pre-Eclamptic Pregnancy
3.1. MIF in Normal Pregnancy
3.1.1. MIF in Early Pregnancy
3.1.2. MIF from Mid-pregnancy to Term
3.1.3. MIF in Fetal–Newborn Blood
3.2. MIF in Pre-Eclampsia
3.2.1. MIF in Women Who Later Develop PE
3.2.2. MIF in Women with Established PE
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur.J. Obs. Gynecol. Reprod. Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef]
- Ghulmiyyah, L.; Sibai, B. Maternal mortality from preeclampsia/eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, J.J.A.; van Heijst, A.F.; de Haan, A.F.J.; van der Heijden, O.W.H. Early-onset preeclampsia is associated with perinatal mortality and severe neonatal morbidity. J. Matern. Fetal Neonatal Med. 2017, 30, 2789–2794. [Google Scholar] [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef]
- ACOG Practice Bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obs. Gynecol. 2002, 99, 159–167. [Google Scholar]
- Rolnik, D.L.; Wright, D.; Poon, L.C.; O’Gorman, N.; Syngelaki, A.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. Aspirin versus placebo in pregnancies at high risk for preterm preeclampsia. N. Engl. J. Med. 2017, 377, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef]
- Quayle, A.J. The innate and early immune response to pathogen challenge in the female genital tract and the pivotal role of epithelial cells. J. Reprod. Immunol. 2002, 57, 61–79. [Google Scholar] [CrossRef]
- Wira, C.R.; Fahey, J.V.; Sentman, C.L.; Pioli, P.A.; Shen, L. Innate and adaptive immunity in female genital tract: Cellular responses and interactions. Immunol. Rev. 2005, 206, 306–335. [Google Scholar] [CrossRef]
- Khan, K.N.; Fujishita, A.; Kitajima, M.; Hiraki, K.; Nakashima, M.; Masuzaki, H. Intra-uterine microbial colonization and occurrence of endometritis in women with endometriosis. Hum. Reprod. 2014, 29, 2446–2456. [Google Scholar] [CrossRef]
- Benjelloun, F.; Quillay, H.; Cannou, C.; Marlin, R.; Madec, Y.; Fernandez, H.; Chrétien, F.; Le Grand, R.; Barré-Sinoussi, F.; Nugeyre, M.T.; et al. Activation of Toll-like receptors differentially modulates inflammation in the human reproductive tract: Preliminary findings. Front. Immunol. 2020, 11, 1655. [Google Scholar] [CrossRef]
- Roach, J.C.; Glusman, G.; Rowen, L.; Kaur, A.; Purcell, M.K.; Smith, K.D.; Hood, L.E.; Aderem, A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9577–9582. [Google Scholar] [CrossRef] [PubMed]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef]
- Abrahams, V.M.; Bole-Aldo, P.; Kim, Y.M.; Straszewski-Chavez, S.L.; Chaiworapongsa, T.; Romero, R.; Mor, G. Divergent trophoblast responses to bacterial products mediated by TLRs. J. Immunol. 2004, 173, 4286–4296. [Google Scholar] [CrossRef]
- Koga, K.; Mor, G. Toll-like receptors at the maternal-fetal interface in normal pregnancy and pregnancy disorders. Am. J. Reprod. Immunol. 2010, 63, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Pudney, J.; He, X.; Masheeb, Z.; Kindelberger, D.W.; Kuohung, W.; Ingalls, R.R. Differential expression of toll-like receptors in the human placenta across early gestation. Placenta 2016, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Ortiz, A.; Flores-Espinosa, P.; Mancilla-Herrera, I.; Vega-Sánchez, R.; Díaz, L.; Zaga-Clavellina, V. Innate immune cells and toll-like receptor-dependent responses at the maternal-fetal interface. Int. J. Mol. Sci. 2019, 20, 3654. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Duriez, M.; Quillay, H.; Madec, Y.; El Costa, H.; Cannou, C.; Marlin, R.; de Truchis, C.; Rahmati, M.; Barré-Sinoussi, F.; Nugeyre, M.T.; et al. Human decidual macrophages and NK cells differentially express Toll-like receptors and display distinct cytokine profiles upon TLR stimulation. Front. Microbiol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Sacks, G.P.; Studena, K.; Sargent, K.; Redman, C.W. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am. J. Obs. Gynecol. 1998, 179, 80–86. [Google Scholar] [CrossRef]
- Abbassi-Ghanavati, M.; Greer, L.G.; Cunningham, F.G. Pregnancy and laboratory studies: A reference table for clinicians. Obs. Gynecol. 2009, 114, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Szarka, A.; Rigó, J.; Lázár, L.; Beko, G.; Molvarec, A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol. 2010, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sacks, G.P.; Sargent, I.L. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am. J. Obs. Gynecol. 1999, 180, 499–506. [Google Scholar] [CrossRef]
- Pineda, A.; Verdin-Terán, S.L.; Camacho, A.; Moreno-Fierros, L. Expression of toll-like receptor TLR-2, TLR-3, TLR-4 and TLR-9 is increased in placentas from patients with preeclampsia. Arch. Med. Res. 2011, 42, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Nizyaeva, N.V.; Kulikova, G.V.; Nagovitsyna, M.N.; Shchegolev, A.I. Peculiarities of the expression of TLR4 and inhibitor of TLR-Cascade tollip in the placenta in earlyand late-onset preeclampsia. Bull. Exp. Biol. Med. 2019, 166, 507–511. [Google Scholar] [CrossRef]
- Zourbas, S.; Dubanchet, S.; Martal, J.; Chaouat, G. Localization of pro-inflammatory (IL-12, IL-15) and anti-inflammatory (IL-11, IL-13) cytokines at the foetomaternal interface during murine pregnancy. Clin. Exp. Immunol. 2001, 126, 519–528. [Google Scholar] [CrossRef]
- Chaouat, G.; Zourbas, S.; Ostojic, S.; Lappree-Delage, G.; Dubanchet, S.; Ledee, N.; Martal, J. A brief review of recent data on some cytokine expressions at the materno-foetal interface which might challenge the classical Th1/Th2 dichotomy. J. Reprod Immunol. 2002, 53, 241–256. [Google Scholar] [CrossRef]
- Mor, G. Pregnancy reconceived. Nat. Hist. 2007, 116, 36–41. [Google Scholar]
- Saito, S.; Nakashima, A.; Shima, T.; Ito, M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am. J. Reprod. Immunol. 2010, 63, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, T.G.; Lin, H.; Guilbert, L.; Mosmann, T.R. Bidirectional cytokine interactions in the maternal-fetal relationship: Is successful pregnancy a TH2 phenomenon? Immunol. Today 1993, 14, 353–356. [Google Scholar] [CrossRef]
- Wegmann, T.G. Foetal protection against abortion: Is it immunosuppression or immunostimulation? Ann. Immunol. 1984, 135, 309–312. [Google Scholar]
- Szekeres-Bartho, J.; Wegmann, T.G. A progesterone-dependent immunomodulatory protein alters the Th1/Th2 balance. J. Reprod. Immunol. 1996, 31, 81–95. [Google Scholar] [CrossRef]
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and pregnancy: The role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef]
- Duc-Goiran, P.; Mignot, T.M.; Bourgeois, C.; Ferré, F. Embryo-maternal interactions at the implantation site: A delicate equilibrium. Eur. J. Obs. Gynecol. Reprod. Biol. 1999, 83, 85–100. [Google Scholar] [CrossRef]
- Dekel, N.; Gnainsky, Y.; Granot, I.; Mor, G. Inflammation and implantation. Am. J. Reprod. Immunol. 2010, 63, 17–21. [Google Scholar] [CrossRef]
- Boeldt, D.S.; Bird, I.M. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J. Endocrinol. 2017, 232, R27–R44. [Google Scholar] [CrossRef]
- Le Bouteiller, P.; Piccinni, M.P. Human NK cells in pregnant uterus: Why there? Am. J. Reprod. Immunol. 2008, 59, 401–406. [Google Scholar] [CrossRef]
- Manaster, I.; Mandelboim, O. The unique properties of uterine NK cells. Am. J. Reprod. Immunol. 2010, 63, 434–444. [Google Scholar] [CrossRef]
- Romero, R.; Espinoza, J.; Gonçalves, L.F.; Kusanovic, J.P.; Friel, L.A.; Nien, J.K. Inflammation in preterm and term labour and delivery. Semin. Fetal Neonatal Med. 2006, 11, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Landon, M.; Galan, H.; Jauniaux, E.; Driscoll, D.; Berghella, V.; Grobman, W.; Kilpatrick, S.; Cahill, A. Gabbe’s Obstetrics: Normal and Problem Pregnancies, 8th ed.; Elsevier: Philadelphia, PA, USA, 2020; p. 1280. [Google Scholar]
- Todros, T.; Bontempo, S.; Piccoli, E.; Ietta, F.; Romagnoli, R.; Biolcati, M.; Castellucci, M.; Paulesu, L. Increased levels of macrophage migration inhibitory factor (MIF) in preeclampsia. Eur. J. Obs. Gynecol. Reprod. Biol. 2005, 123, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, A.; Giuffrida, D.; Nuzzo, A.M.; Pierobon, D.; Cardaropoli, S.; Piccoli, E.; Giovarelli, M.; Todros, T. Pro-inflammatory profile of preeclamptic placental mesenchymal stromal cells: New insights into the etiopathogenesis of preeclampsia. PLoS ONE 2013, 8, e59403. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, D.; Cetin, I.; Frusca, T.; Ferrazzi, E.; Fuse’, F.; Gervasi, M.T.; Plebani, M.; Todros, T. Italian advisory board: sFlt-1/PlGF ratio and preeclampsia, state of the art and developments in diagnostic, therapeutic and clinical management. Eur. J. Obs. Gynecol. Reprod. Biol. 2016, 206, 70–73. [Google Scholar] [CrossRef]
- Morano, D.; Rolfo, A.; Tisato, V.; Farina, A.; Rimondi, E.; Scutiero, G.; Greco, P.; Bonaccorsi, G.; Todros, T. Lower maternal serum tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) levels in early preeclampsia. A retrospective study. Pregnancy Hypertens. 2018, 12, 1–5. [Google Scholar] [CrossRef]
- Huppertz, B.; Kingdom, J.; Caniggia, I.; Desoye, G.; Black, S.; Korr, H.; Kaufmann, P. Hypoxia favours necrotic versus apoptotic shedding of placental syncytiotrophoblast into the maternal circulation. Placenta 2003, 24, 181–190. [Google Scholar] [CrossRef]
- Ponzetto, A.; Cardaropoli, S.; Piccoli, E.; Rolfo, A.; Gennero, L.; Kanduc, D.; Todros, T. Pre-eclampsia is associated with Helicobacter pylori seropositivity in Italy. J. Hypertens. 2006, 24, 2445–2449. [Google Scholar] [CrossRef]
- Todros, T.; Vasario, E.; Cardaropoli, S. Preeclampsia as an infectious disease. Expert Rev. Obstet. Gynecol. 2007, 2, 735–741. [Google Scholar] [CrossRef]
- Kell, D.B.; Kenny, L.C. A dormant microbial component in the development of preeclampsia. Front. Med. 2016, 3, 60. [Google Scholar] [CrossRef]
- Di Simone, N.; Tersigni, C.; Cardaropoli, S.; Franceschi, F.; Di Nicuolo, F.; Castellani, R.; Bugli, F.; de Waure, C.; Cavaliere, A.F.; Gasbarrini, A.; et al. Helicobacter pylori infection contributes to placental impairment in preeclampsia: Basic and clinical evidences. Helicobacter 2017, 22, 12347. [Google Scholar] [CrossRef] [PubMed]
- Cardaropoli, S.; Todros, T.; Nuzzo, A.M.; Rolfo, A. Maternal serum levels and placental expression of hepcidin in preeclampsia. Pregnancy Hypertens. 2018, 11, 47–53. [Google Scholar] [CrossRef]
- Gyselaers, W. Preeclampsia is a syndrome with a cascade of pathophysiologic events. J. Clin. Med. 2020, 9, 2245. [Google Scholar] [CrossRef] [PubMed]
- Todros, T.; Masturzo, B.; De Francia, S. COVID-19 infection: ACE2, pregnancy and preeclampsia. Eur. J. Obs. Gynecol. Reprod. Biol. 2020, 253, 330. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L.; Staff, A.C. IFPA senior award lecture: Making sense of pre-eclampsia–Two placental causes of preeclampsia? Placenta 2014, 35, S20–S25. [Google Scholar] [CrossRef]
- Marzioni, D.; Todros, T.; Cardaropoli, S.; Rolfo, A.; Lorenzi, T.; Ciarmela, P.; Romagnoli, R.; Paulesu, L.; Castellucci, M. Activating protein-1 family of transcription factors in the human placenta complicated by preeclampsia with and without fetal growth restriction. Placenta 2010, 31, 919–927. [Google Scholar] [CrossRef]
- Cardaropoli, S.; Rolfo, A.; Piazzese, A.; Ponzetto, A.; Todros, T. Helicobacter pylori’s virulence and infection persistence define pre-eclampsia complicated by fetal growth retardation. World J. Gastroenterol. 2011, 17, 5156–5165. [Google Scholar] [CrossRef]
- Ferrazzi, E.; Zullino, S.; Stampalija, T.; Vener, C.; Cavoretto, P.; Gervasi, M.T.; Vergani, P.; Mecacci, F.; Marozio, L.; Oggè, G.; et al. Bedside diagnosis of two major clinical phenotypes of hypertensive disorders of pregnancy. Ultrasound Obs. Gynecol. 2016, 48, 224–231. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Immunology of pre-eclampsia. Am. J. Repro Immuno 2010, 63, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Todros, T.; Sciarrone, A.; Piccoli, E.; Guiot, C.; Kaufmann, P.; Kingdom, J. Umbilical doppler waveforms and placental villous angiogenesis in pregnancies complicated by fetal growth restriction. Obs. Gynecol. 1999, 93, 499–503. [Google Scholar]
- Kingdom, J.; Huppertz, B.; Seaward, G.; Kaufmann, P. Development of the placental villous tree and its consequences for fetal growth. Eur J. Obs. Gynecol. Reprod. Biol. 2000, 92, 35–43. [Google Scholar] [CrossRef]
- Salafia, C.M.; Charles, A.K.; Maas, E.M. Placenta and fetal growth restriction. Clin. Obs. Gynecol. 2006, 49, 236–256. [Google Scholar] [CrossRef]
- Maynard, S.; Epstein, F.H.; Karumanchi, S.A. Preeclampsia and angiogenic imbalance. Annu. Rev. Med. 2008, 59, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Ness, R.B.; Roberts, J.M. Heterogeneous causes constituting the single syndrome of preeclampsia: A hypothesis and its implications. Am. J. Obs. Gynecol. 1996, 175, 1365–1370. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef]
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966, 153, 80–82. [Google Scholar] [CrossRef]
- David, J.R. Delayed hypersensitivity in vitro: Its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc. Natl. Acad. Sci. USA 1966, 56, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; VanPatten, S.; Deen, N.S.; Al-Abed, Y.; Morand, E.F. Rediscovering MIF: New tricks for an old cytokine. Trends Immunol. 2019, 40, 447–462. [Google Scholar] [CrossRef]
- Petrovsky, N.; Socha, L.; Silva, D.; Grossman, A.B.; Metz, C.; Bucala, R. Macrophage migration inhibitory factor exhibits a pronounced circadian rhythm relevant to its role as a glucocorticoid counter-regulator. Immunol. Cell Biol. 2003, 81, 137–143. [Google Scholar] [CrossRef]
- Vincent, F.B.; Lin, E.; Sahhar, J.; Ngian, G.S.; Kandane-Rathnayake, R.; Mende, R.; Hoi, A.Y.; Morand, E.F.; Lang, T.; Harris, J. Analysis of serum macrophage migration inhibitory factor and D-dopachrome tautomerase in systemic sclerosis. Clin. Transl. Immunol. 2018, 7, e1042. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.A.; Metz, C.N.; Peng, T.; Bucala, R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J. Biol. Chem. 1999, 274, 18100–18106. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Rosengren, E.; Aman, P.; Thelin, S.; Hansson, C.; Ahlfors, S.; Björk, P.; Jacobsson, L.; Rorsman, H. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett 1997, 417, 85–88. [Google Scholar] [CrossRef]
- Kleemann, R.; Kapurniotu, A.; Frank, R.W.; Gessner, A.; Mischke, R.; Flieger, O.; Jüttner, S.; Brunner, H.; Bernhagen, J. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J. Mol. Biol 1998, 280, 85–102. [Google Scholar] [CrossRef]
- Bucala, R.; Donnelly, S.C. Macrophage migration inhibitory factor: A probable link between inflammation and cancer. Immunity 2007, 26, 281–285. [Google Scholar] [CrossRef]
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef]
- Cunha, F.Q.; Weiser, W.Y.; David, J.R.; Moss, D.W.; Moncada, S.; Liew, F.Y. Recombinant migration inhibitory factor induces nitric oxide synthase in murine macrophages. J. Immunol. 1993, 150, 1908–1912. [Google Scholar]
- Calandra, T.; Bernhagen, J.; Metz, C.N.; Spiegel, L.A.; Bacher, M.; Donnelly, T.; Cerami, A.; Bucala, R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature 1995, 377, 68–71. [Google Scholar] [CrossRef]
- Reyes, J.L.; Terrazas, L.I.; Espinoza, B.; Cruz-Robles, D.; Soto, V.; Rivera-Montoya, I.; Gómez-García, L.; Snider, H.; Satoskar, A.R.; Rodríguez-Sosa, M. Macrophage migration inhibitory factor contributes to host defense against acute Trypanosoma cruzi infection. Infect. Immun. 2006, 74, 3170–3179. [Google Scholar] [CrossRef]
- Arjona, A.; Foellmer, H.G.; Town, T.; Leng, L.; McDonald, C.; Wang, T.; Wong, S.J.; Montgomery, R.R.; Fikrig, E.; Bucala, R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J. Clin. Investig. 2007, 117, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Ferro, E.A.; Mineo, J.R.; Ietta, F.; Bechi, N.; Romagnoli, R.; Silva, D.A.; Sorda, G.; Bevilacqua, E.; Paulesu, L.R. Macrophage migration inhibitory factor is up-regulated in human first-trimester placenta stimulated by soluble antigen of Toxoplasma gondii, resulting in increased monocyte adhesion on villous explants. Am. J. Pathol. 2008, 172, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Delaloye, J.; Chanson, A.L.; Giddey, M.; Le Roy, D.; Calandra, T. Macrophage migration inhibitory factor deficiency is associated with impaired killing of gram-negative bacteria by macrophages and increased susceptibility to Klebsiella pneumoniae sepsis. J. Infect. Dis 2013, 207, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, I.; Mirkov, I.; Kataranovski, M.; Glamoclija, J.; Stosic-Grujicic, S. A role for macrophage migration inhibitory factor in protective immunity against Aspergillus fumigatus. Immunobiology 2011, 216, 1018–1027. [Google Scholar] [CrossRef]
- Roger, T.; David, J.; Glauser, M.P.; Calandra, T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 2001, 414, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Roger, T.; Froidevaux, C.; Martin, C.; Calandra, T. Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). J. Endotoxin Res. 2003, 9, 119–123. [Google Scholar] [CrossRef]
- Arizza, V.; Bonura, A.; La Paglia, L.; Urso, A.; Pinsino, A.; Vizzini, A. Transcriptional and in silico analyses of MIF cytokine and TLR signalling interplay in the LPS inflammatory response of Ciona robusta. Sci. Rep. 2020, 10, 11339. [Google Scholar] [CrossRef]
- Bilsborrow, J.B.; Doherty, E.; Tilstam, P.V.; Bucala, R. Macrophage migration inhibitory factor (MIF) as a therapeutic target for rheumatoid arthritis and systemic lupus erythematosus. Expert Opin. Targets 2019, 23, 733–744. [Google Scholar] [CrossRef]
- Grieb, G.; Merk, M.; Bernhagen, J.; Bucala, R. Macrophage migration inhibitory factor (MIF): A promising biomarker. Drug News Perspect. 2010, 23, 257–264. [Google Scholar] [CrossRef]
- Hertelendy, J.; Reumuth, G.; Simons, D.; Stoppe, C.; Kim, B.S.; Stromps, J.P.; Fuchs, P.C.; Bernhagen, J.; Pallua, N.; Grieb, G. Macrophage migration inhibitory factor—A favorable marker in inflammatory diseases? Curr. Med. Chem. 2018, 25, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Mikulowska, A.; Metz, C.N.; Bucala, R.; Holmdahl, R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J. Immunol. 1997, 158, 5514–5517. [Google Scholar]
- Calandra, T.; Echtenacher, B.; Roy, D.L.; Pugin, J.; Metz, C.N.; Hültner, L.; Heumann, D.; Männel, D.; Bucala, R.; Glauser, M.P. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat. Med. 2000, 6, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Nishihira, J.; Miki, I. Blockade of macrophage migration inhibitory factor (MIF) prevents the antigen-induced response in a murine model of allergic airway inflammation. Inflamm. Res. 2007, 56, 24–31. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, X.; Li, W.; Shan, C.; Song, J.; Zhang, M. Inhibition of macrophage migration inhibitory factor protects against inflammation through a toll-like receptor-related pathway after diffuse axonal injury in rats. Biomed. Res. Int. 2020, 2020, 5946205. [Google Scholar] [CrossRef]
- Cavalli, E.; Ciurleo, R.; Petralia, M.C.; Fagone, P.; Bella, R.; Mangano, K.; Nicoletti, F.; Bramanti, P.; Basile, M.S. Emerging role of the macrophage migration inhibitory factor family of cytokines in neuroblastoma. pathogenic effectors and novel therapeutic targets? Molecules 2020, 25, 1194. [Google Scholar] [CrossRef] [PubMed]
- Paulesu, L.; Bhattacharjee, J.; Bechi, N.; Romagnoli, R.; Jantra, S.; Ietta, F. Pro-inflammatory cytokines in animal and human gestation. Curr. Pharm. Des. 2010, 16, 3601–3615. [Google Scholar] [CrossRef]
- Viganò, P.; Cintorino, M.; Schatz, F.; Lockwood, C.J.; Arcuri, F. The role of macrophage migration inhibitory factor in maintaining the immune privilege at the fetal-maternal interface. Semin. Immunopathol. 2007, 29, 135–150. [Google Scholar] [CrossRef]
- Bevilacqua, E.; Paulesu, L.; Ferro, E.A.; Ietta, F.; Faria, M.R.; Lorenzon, A.R.; Costa, A.F.; Martucci, M. Review: Putative roles for the macrophage migratory inhibitory factor at the maternal fetal interface. Placenta 2014, 35, S51–S56. [Google Scholar] [CrossRef]
- Fingerle-Rowson, G.; Petrenko, O.; Metz, C.N.; Forsthuber, T.G.; Mitchell, R.; Huss, R.; Moll, U.; Müller, W.; Bucala, R. The p53–dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. USA 2003, 100, 9354–9359. [Google Scholar] [CrossRef]
- Ietta, F.; Todros, T.; Ticconi, C.; Piccoli, E.; Zicari, A.; Piccione, E.; Paulesu, L. Macrophage migration inhibitory factor in human pregnancy and labor. Am. J. Reprod. Immunol. 2002, 48, 404–409. [Google Scholar] [CrossRef]
- Pearce, B.D.; Garvin, S.E.; Grove, J.; Bonney, E.A.; Dudley, D.J.; Schendel, D.E.; Thorsen, P. Serum macrophage migration inhibitory factor in the prediction of preterm delivery. Am. J. Obs. Gynecol. 2008, 199, 46.e1–46.e6. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, F.; Ricci, C.; Ietta, F.; Cintorino, M.; Tripodi, S.A.; Cetin, I.; Garzia, E.; Schatz, F.; Klemi, P.; Santopietro, R.; et al. Macrophage migration inhibitory factor in the human endometrium: Expression and localization during the menstrual cycle and early pregnancy. Biol. Reprod. 2001, 64, 1200–1205. [Google Scholar] [CrossRef]
- Kats, R.; Al-Akoum, M.; Guay, S.; Metz, C.; Akoum, A. Cycle-dependent expression of macrophage migration inhibitory factor in the human endometrium. Hum. Reprod. 2005, 20, 3518–3525. [Google Scholar] [CrossRef]
- Arcuri, F.; Cintorino, M.; Carducci, A.; Papa, S.; Riparbelli, M.G.; Mangioni, S.; Di Blasio, A.M.; Tosi, P.; Viganò, P. Human decidual natural killer cells as a source and target of macrophage migration inhibitory factor. Reproduction 2006, 131, 175–182. [Google Scholar] [CrossRef]
- Arcuri, F.; Cintorino, M.; Vatti, R.; Carducci, A.; Liberatori, S.; Paulesu, L. Expression of macrophage migration inhibitory factor transcript and protein by first-trimester human trophoblasts. Biol. Reprod. 1999, 60, 1299–1303. [Google Scholar] [CrossRef]
- Ietta, F.; Wu, Y.; Romagnoli, R.; Soleymanlou, N.; Orsini, B.; Zamudio, S.; Paulesu, L.; Caniggia, I. Oxygen regulation of macrophage migration inhibitory factor in human placenta. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E272–E280. [Google Scholar] [CrossRef]
- Jovanović Krivokuća, M.; Stefanoska, I.; Abu Rabi, T.; Al-Abed, Y.; Stošić-Grujičić, S.; Vićovac, L. Pharmacological inhibition of MIF interferes with trophoblast cell migration and invasiveness. Placenta 2015, 36, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ietta, F.; Ferro, E.A.V.; Bevilacqua, E.; Benincasa, L.; Maioli, E.; Paulesu, L. Role of the macrophage Migration Inhibitory Factor (MIF) in the survival of first trimester human placenta under induced stress conditions. Sci. Rep. 2018, 8, 12150. [Google Scholar] [CrossRef]
- Vilotic, A.; Jovanovic Krivokuca, M.; Stefanoska, I.; Vrzic Petronijevic, S.; Petronijevic, M.; Vicovac, L. Macrophage migration inhibitory factor is involved in endovascular trophoblast cell function. EXCLI J. 2019, 18, 1007. [Google Scholar]
- Zicari, A.; Ticconi, C.; Ietta, F.; Belmonte, A.; Bechi, N.; Realacci, M.; Di Vito, M.; Arcuri, F.; Russo, M.; Piccione, E.; et al. Macrophage migration inhibitory factor-nitric oxide interaction in human fetal membranes at term pregnancy. J. Soc. Gynecol. Investig. 2006, 13, 263–270. [Google Scholar] [CrossRef]
- Yamada, H.; Kato, E.H.; Morikawa, M.; Shimada, S.; Saito, H.; Watari, M.; Minakami, H.; Nishihira, J. Decreased serum levels of macrophage migration inhibition factor in miscarriages with normal chromosome karyotype. Hum. Reprod. 2003, 18, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Comba, C.; Bastu, E.; Dural, O.; Yasa, C.; Keskin, G.; Ozsurmeli, M.; Buyru, F.; Serdaroglu, H. Role of inflammatory mediators in patients with recurrent pregnancy loss. Fertil. Steril. 2015, 104, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Cardaropoli, S.; Paulesu, L.; Romagnoli, R.; Ietta, F.; Marzioni, D.; Castellucci, M.; Rolfo, A.; Vasario, E.; Piccoli, E.; Todros, T. Macrophage migration inhibitory factor in fetoplacental tissues from preeclamptic pregnancies with or without fetal growth restriction. Clin. Dev. Immunol. 2012, 2012, 639342. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Nasri, H.; Nasr, A.M.; Adam, I. Maternal and umbilical cord blood level of macrophage migration inhibitory factor and insulin like growth factor in Sudanese women with preeclampsia. J. Obs. Gynaecol. 2019, 39, 63–67. [Google Scholar] [CrossRef]
- Galbiati, S.; Inversetti, A.; Causarano, V.; Stenirri, S.; Soriani, N.; Ambrosi, A.; Valsecchi, L.; Candiani, M.; Cremonesi, L.; Ferrari, M.; et al. HIF1A and MIF as potential predictive mRNA biomarkers of pre-eclampsia: A longitudinal prospective study in high risk population. Clin. Chem. Lab. Med. 2015, 53, 1339–1347. [Google Scholar] [CrossRef]
- Hristoskova, S.; Holzgreve, W.; Zhong, X.Y.; Hahn, S. Macrophage migration inhibition factor is elevated in pregnancy, but not to a greater extent in preeclampsia. Arch. Gynecol. Obs. 2006, 274, 25–28. [Google Scholar] [CrossRef][Green Version]
- Cardaropoli, S.; Ietta, F.; Romagnoli, R.; Rolfo, A.; Paulesu, L.; Todros, T. Lower macrophage migration inhibitory factor concentrations in maternal serum before pre-eclampsia onset. J. Interferon Cytokine Res. 2014, 34, 537–542. [Google Scholar] [CrossRef]
- Graham, C.H.; Hawley, T.S.; Hawley, R.G.; MacDougall, J.R.; Kerbel, R.S.; Khoo, N.; Lala, P.K. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp. Cell Res. 1993, 206, 204–211. [Google Scholar] [CrossRef]
- Jovanović-Krivokuća, M.; Stefanoska, I.; Abu Rabi, T.; Vilotić, A.; Petronijević, M.; Vrzić-Petronijević, S.; Radojčić, L.; Vićovac, L. MIF is among the proinflammatory cytokines increased by LPS in the human trophoblast line. Arch. Biol. Sci. 2016, 68, 715–722. [Google Scholar] [CrossRef]
- Sharp, A.N.; Heazell, A.E.; Crocker, I.P.; Mor, G. Placental apoptosis in health and disease. Am. J. Reprod. Immunol. 2010, 64, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Przybyl, L.; Haase, N.; Golic, M.; Rugor, J.; Solano, M.E.; Arck, P.C.; Gauster, M.; Huppertz, B.; Emontzpohl, C.; Stoppe, C.; et al. CD74-Downregulation of placental macrophage-trophoblastic interactions in preeclampsia. Circ. Res. 2016, 119, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.F.; Gomes, S.Z.; Lorenzon-Ojea, A.R.; Martucci, M.; Faria, M.R.; Pinto, D.S.; Oliveira, S.F.; Ietta, F.; Paulesu, L.; Bevilacqua, E. Macrophage migration inhibitory factor induces phosphorylation of Mdm2 mediated by phosphatidylinositol 3-kinase/Akt kinase: Role of this pathway in decidual cell survival. Placenta 2016, 41, 27–38. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef]
- Roger, T.; Schneider, A.; Weier, M.; Sweep, F.C.; Le Roy, D.; Bernhagen, J.; Calandra, T.; Giannoni, E. High expression levels of macrophage migration inhibitory factor sustain the innate immune responses of neonates. Proc. Natl. Acad. Sci. USA 2016, 113, E997–E1005. [Google Scholar] [CrossRef]
- Roger, T.; Schlapbach, L.J.; Schneider, A.; Weier, M.; Wellmann, S.; Marquis, P.; Vermijlen, D.; Sweep, F.C.; Leng, L.; Bucala, R.; et al. Plasma levels of macrophage migration inhibitory factor and d-dopachrome tautomerase show a highly specific profile in early life. Front. Immunol. 2017, 8, 26. [Google Scholar] [CrossRef]
- Chaisavaneeyakorn, S.; Moore, J.M.; Othoro, C.; Otieno, J.; Chaiyaroj, S.C.; Shi, Y.P.; Nahlen, B.L.; Lal, A.A.; Udhayakumar, V. Immunity to placental malaria. IV. Placental malaria is associated with up-regulation of macrophage migration inhibitory factor in intervillous blood. J. Infect. Dis. 2002, 186, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Vera, P.L.; Meyer-Siegler, K.L. Inflammation of the rat prostate evokes release of macrophage migration inhibitory factor in the bladder: Evidence for a viscerovisceral reflex. J. Urol. 2004, 172, 2440–2445. [Google Scholar] [CrossRef]
- Arndt, U.; Wennemuth, G.; Barth, P.; Nain, M.; Al-Abed, Y.; Meinhardt, A.; Gemsa, D.; Bacher, M. Release of macrophage migration inhibitory factor and CXCL8/interleukin-8 from lung epithelial cells rendered necrotic by influenza A virus infection. J. Virol. 2002, 76, 9298–9306. [Google Scholar] [CrossRef]
- Chaisavaneeyakorn, S.; Lucchi, N.; Abramowsky, C.; Othoro, C.; Chaiyaroj, S.C.; Shi, Y.P.; Nahlen, B.L.; Peterson, D.S.; Moore, J.M.; Udhayakumar, V. Immunohistological characterization of macrophage migration inhibitory factor expression in Plasmodium falciparum-infected placentas. Infect. Immun. 2005, 73, 3287–3293. [Google Scholar] [CrossRef]



{kind=link}
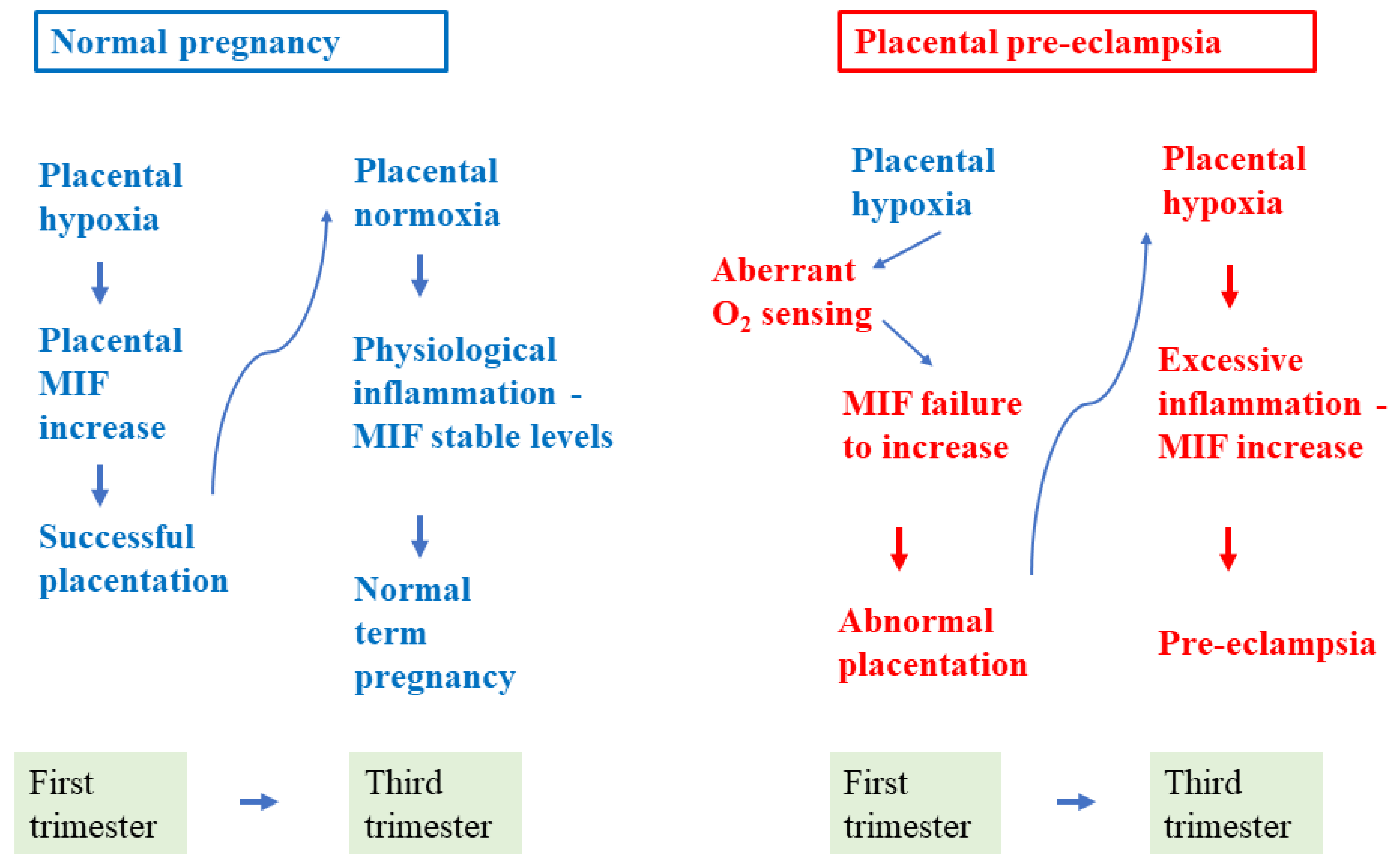{kind=link}
| Findings | References | |
|---|---|---|
| Menstrual cycle | MIF mRNA and protein are expressed in uterine glandular and surface epithelium. | [104] |
| Uterine MIF expression is higher in late proliferative and secretory phases. | [105] | |
| Early pregnancy | MIF is produced by uNK cells and acts on these same cells by reducing their cytolytic activity. | [106] |
| MIF protein is expressed in first trimester placenta mainly in villous and extravillous trophoblast. | [107] | |
| Trophoblast MIF is induced by hypoxia. | [108] | |
| MIF promotes trophoblast cell invasion and migration. | [109] | |
| MIF promotes survival of first trimester human placenta under induced stress conditions. | [110] | |
| MIF promotes trophoblast differentiation to endovascular phenotype. | [111] | |
| Mid-pregnancy | MIF mRNA and protein placenta expression declines at 11–12 weeks and remains stable until term. | [108] |
| Term pregnancy | MIF in amniotic fluid is higher at term than at mid-gestation and higher at term with spontaneous delivery. | [102] |
| MIF levels in umbilical cord serum at term birth are higher than in maternal serum. | [102] | |
| MIF is expressed and secreted by extraembryonic membranes. | [112] |
| Findings | References | |
|---|---|---|
| Miscarriage | Maternal serum MIF levels in early pregnancy are low in patients having miscarriage. | [113] |
| MIF in uterine tissues and maternal blood is low in patients with recurrent pregnancy loss. | [114] | |
| Pre-term delivery | Maternal plasma MIF levels at first–second trimester are higher in pregnancies with preterm delivery. | [103] |
| Pre-eclampsia | Maternal serum MIF levels are higher in patients with PE than in normal pregnancy. | [115,116] |
| Maternal serum MIF is higher in patients affected by IUGR-PE while not in AGA-PE. | [115] | |
| MIF mRNA in maternal plasma at 24–30 weeks is higher in patients who later develop PE. | [117] | |
| MIF maternal serum levels are higher in normal pregnancy compared to non-pregnancy but not further increased in PE patients. | [118] | |
| Secretion of MIF by placental mesenchymal stromal cells is higher in IUGR-PE than in normal pregnancy. | [45] | |
| Maternal serum MIF in first–early second trimester is lower in women who later develop PE. | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todros, T.; Paulesu, L.; Cardaropoli, S.; Rolfo, A.; Masturzo, B.; Ermini, L.; Romagnoli, R.; Ietta, F. Role of the Macrophage Migration Inhibitory Factor in the Pathophysiology of Pre-Eclampsia. Int. J. Mol. Sci. 2021, 22, 1823. https://doi.org/10.3390/ijms22041823
Todros T, Paulesu L, Cardaropoli S, Rolfo A, Masturzo B, Ermini L, Romagnoli R, Ietta F. Role of the Macrophage Migration Inhibitory Factor in the Pathophysiology of Pre-Eclampsia. International Journal of Molecular Sciences. 2021; 22(4):1823. https://doi.org/10.3390/ijms22041823
Chicago/Turabian StyleTodros, Tullia, Luana Paulesu, Simona Cardaropoli, Alessandro Rolfo, Bianca Masturzo, Leonardo Ermini, Roberta Romagnoli, and Francesca Ietta. 2021. "Role of the Macrophage Migration Inhibitory Factor in the Pathophysiology of Pre-Eclampsia" International Journal of Molecular Sciences 22, no. 4: 1823. https://doi.org/10.3390/ijms22041823
APA StyleTodros, T., Paulesu, L., Cardaropoli, S., Rolfo, A., Masturzo, B., Ermini, L., Romagnoli, R., & Ietta, F. (2021). Role of the Macrophage Migration Inhibitory Factor in the Pathophysiology of Pre-Eclampsia. International Journal of Molecular Sciences, 22(4), 1823. https://doi.org/10.3390/ijms22041823