
Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
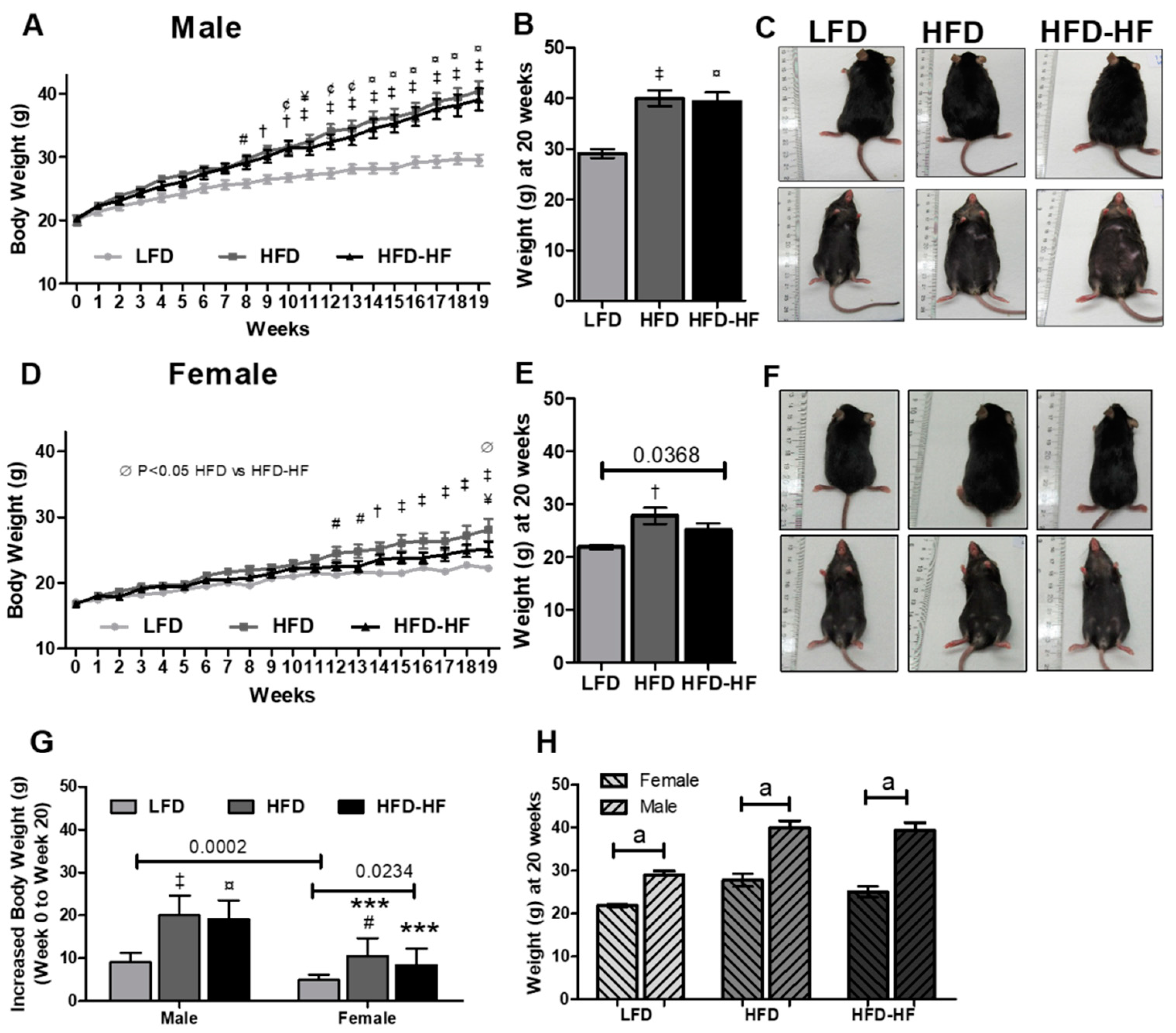
2.1. Effect of Western Diets on Body Weight and Food Consumption
2.2. Altered Fasting Blood Glucose on Western Diet
2.3. Increased Liver Weight under Western Diet Exposure
2.4. Altered Glucose Tolerance and Insulin Sensitivity under Western Diets Exposure
2.5. Altered Liver Regulatory Markers under Western Diets Exposure
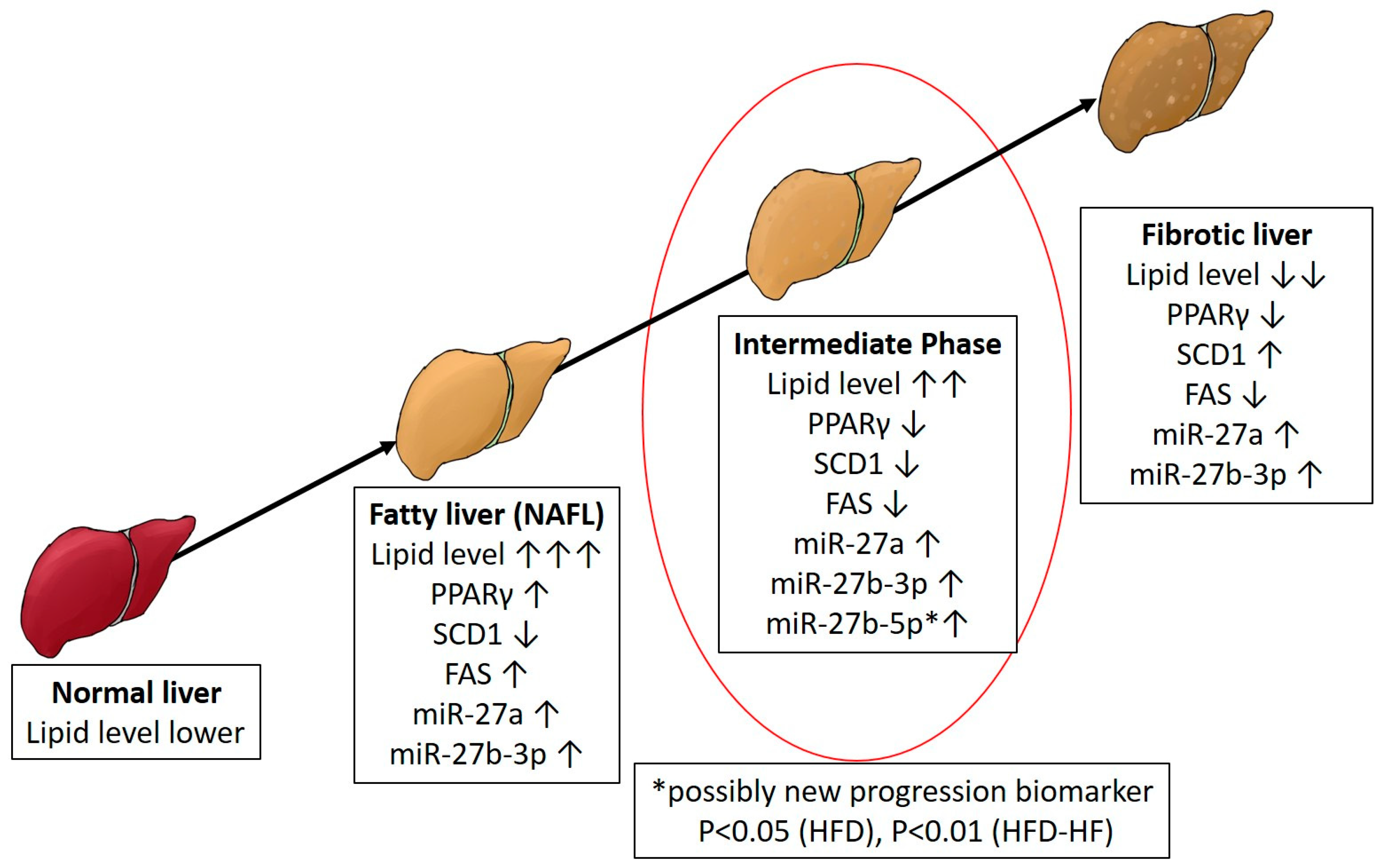
2.6. Altered MicroRNA-27 Expression under Western Diets Exposure
2.7. Fibrosis Markers under Western Diets Exposure
2.8. Lipid Accumulation under Western Diets
3. Discussion
4. Material and Methods
4.1. Mice
4.2. Body Weight and Fasting Blood Glucose
4.3. Glucose and Insulin Tolerance Tests
4.4. miRNA Real Time PCR
4.5. mRNA Real Time RT-PCR
4.6. Western Blot Analysis
4.7. Liver Tissue Histological Analysis
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- William Herman, P.Z. Type 2 Diabetes: An Epidemic Requiring Global Attention and Urgent Action. Diabetes Care 2012, 35, 943–944. [Google Scholar] [CrossRef]
- Gotto, A.M., Jr.; Blackburn, G.L.; Dailey, G.E., 3rd; Garber, A.J.; Grundy, S.M.; Sobel, B.E.; Weir, M.R. The metabolic syndrome: A call to action. Coron. Artery Dis. 2006, 17, 77–80. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef]
- Hagstrom, H.; Elfwen, O.; Hultcrantz, R.; Stal, P. Steatohepatitis is not Associated with an Increased Risk for Fibrosis Progression in Nonalcoholic Fatty Liver Disease. Gastroenterol. Res. Pract. 2018, 2018, 1942648. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Stal, P. Liver fibrosis in non-alcoholic fatty liver disease-diagnostic challenge with prognostic significance. World J. Gastroenterol. 2015, 21, 11077–11087. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V.; Group, L.S. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.; Bischoff, S.C.; Bergheim, I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptors in liver disease. Hepatology 2011, 53, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.K.S.; Peixoto, C.A. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell. Mol. Life Sci. 2018, 75, 2951–2961. [Google Scholar] [CrossRef]
- Gazit, V.; Huang, J.; Weymann, A.; Rudnick, D.A. Analysis of the Role of Hepatic PPARg Expression during Mouse Liver Regeneration. Hepatology 2012, 56, 1489–1498. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor gamma. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef]
- Zeng, X.Y.; Zhang, Y.Q.; He, X.M.; Wan, L.Y.; Wang, H.; Ni, Y.R.; Wang, J.; Wu, J.F.; Liu, C.B. Suppression of hepatic stellate cell activation through downregulation of gremlin1 expression by the miR-23b/27b cluster. Oncotarget 2016, 7, 86198–86210. [Google Scholar] [CrossRef][Green Version]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; Horcel, L.D.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-Induced Hepatocyte-Derived Extracellular Vesicles Regulate Hepatic Stellate Cells via MicroRNA Targeting Peroxisome Proliferator-Activated Receptor-g. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Eguchi, A.; Li, H.; Johnson, C.D.; Papouchado, B.G.; Wree, A.; Messer, K.; Feldstein, A.E. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS ONE 2014, 9, e113651. [Google Scholar] [CrossRef]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Chango, A.; Pogribny, I.P. Considering maternal dietary modulators for epigenetic regulation and programming of the fetal epigenome. Nutrients 2015, 7, 2748–2770. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Krishna, V.; Chatterjee, K.; Tsukamoto, H. Peroxisome proliferatoractivated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Yan, J.; Jiang, J.; Zhou, X.; Lu, Y.; Jiang, H. MicroRNA-27a-3p suppression of peroxisome proliferator-activated receptor-gamma contributes to cognitive impairments resulting from sevoflurane treatment. J. Neurochem. 2017, 143, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. MicroRNAs in Nonalcoholic Fatty Liver Disease. J. Clin. Med. 2015, 4, 1977–1988. [Google Scholar] [CrossRef]
- Mubaid, S.; Ma, J.F.; Omer, A.; Ashour, K.; Lian, X.J.; Sanchez, B.J.; Robinson, S.; Cammas, A.; Dormoy-Raclet, V.; Di Marco, S.; et al. HuR counteracts miR-330 to promote STAT3 translation during inflammation-induced muscle wasting. Proc. Natl. Acad. Sci. USA 2019, 116, 17261–17270. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef]
- Le Lu, J.W.; Lu, H.; Zhang, G.; Liu, Y.; Wang, J.; Zhang, Y.; Shang, H.; Ji, H.; Chen, X.; Duan, Y.; et al. MicroRNA-130a and -130b enhance activation of hepatic stellate cells by suppressing PPARg expression: A rat fibrosis model study. Biochem. Biophys. Res. Commun. 2015, 465, 387–393. [Google Scholar] [CrossRef]
- Bouvy-Liivrand, M.; Heinäniemi, M.; John, E.; Schneider, J.G.; Sauter, T.; Sinkkonen, L. Combinatorial regulation of lipoprotein lipase by microRNAs during mouse adipogenesis. RNA Biol. 2014, 11, 76–91. [Google Scholar] [CrossRef]
- Kulyte, A.; Kwok, K.H.M.; de Hoon, M.; Carninci, P.; Hayashizaki, Y.; Arner, P.; Arner, E. MicroRNA-27a/b-3p and PPARG regulate SCAMP3 through a feed-forward loop during adipogenesis. Sci. Rep. 2019, 9, 13891. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego-Duran, R.; Gallego, P.; Grande, L. Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Chukijrungroat, N.; Khamphaya, T.; Weerachayaphorn, J.; Songserm, T.; Saengsirisuwan, V. Hepatic FGF21 mediates sex differences in high-fat high-fructose diet-induced fatty liver. Am. J. Physiol. Endocr. Metab. 2017, 313, E203–E212. [Google Scholar] [CrossRef]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef] [PubMed]
- Medrikova, D.; Jilkova, Z.M.; Bardova, K.; Janovska, P.; Rossmeisl, M.; Kopecky, J. Sex differences during the course of diet-induced obesity in mice: Adipose tissue expandability and glycemic control. Int. J. Obes. 2012, 36, 262–272. [Google Scholar] [CrossRef]
- Bauer, S.A.; Arndt, T.P.; Leslie, K.E.; Pearl, D.L.; Turner, P.V. Obesity in rhesus and cynomolgus macaques: A comparative review of the condition and its implications for research. Comp. Med. 2011, 61, 514–526. [Google Scholar]
- Inui, A. Obesity—A chronic health problem in cloned mice? Trends Pharmacol. Sci. 2003, 24, 77–80. [Google Scholar] [CrossRef]
- Pellizzon, M.A.; Ricci, M.R. The common use of improper control diets in diet-induced metabolic disease research confounds data interpretation: The fiber factor. Nutr. Metab. 2018, 15, 3. [Google Scholar] [CrossRef]
- Collins, F.S.; Tabak, L.K. NIH calls for increased rigor and reproducibility. Nature 2014, 2014, 612–613. [Google Scholar] [CrossRef]
- Nizar, J.M.; Dong, W.; McClellan, R.B.; Labarca, M.; Zhou, Y.; Wong, J.; Goens, D.G.; Zhao, M.; Velarde, N.; Bernstein, D.; et al. Na+-sensitive elevation in blood pressure is ENaC independent in diet-induced obesity and insulin resistance. Am. J. Physiol. Ren. Physiol. 2016, 310, F812–F820. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Sievenpiper, J.L.; de Souza, R.J.; Cozma, A.I.; Mirrahimi, A.; Carleton, A.J.; Ha, V.; Di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2014, 68, 416–423. [Google Scholar] [CrossRef]
- Macdonald, I.A. A review of recent evidence relating to sugars, insulin resistance and diabetes. Eur. J. Nutr. 2016, 55, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Skat-Rordam, J.; Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor gamma in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, H.F., Jr.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef] [PubMed]
- Peyrou, M.; Ramadori, P.; Bourgoin, L.; Foti, M. PPARs in Liver Diseases and Cancer: Epigenetic Regulation by MicroRNAs. PPAR Res. 2012, 2012, 757803. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, W.; Zhou, M.; Tang, Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci. Rep. 2017, 7, 14493. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef]
- Dorn, C.; Riener, M.O.; Kirovski, G.; Saugspier, M.; Steib, K.; Weiss, T.S.; Gabele, E.; Kristiansen, G.; Hartmann, A.; Hellerbrand, C. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int. J. Clin. Exp. Pathol. 2010, 3, 505–514. [Google Scholar]
- Gianotti, T.F.; Burgueño, A.; Mansilla, N.G.; Pirola, C.J.; Sookoian, S. Fatty liver is associated with transcriptional downregulation of stearoyl-CoA desaturase and impaired protein dimerization. PLoS ONE 2013, 8, e76912. [Google Scholar]
- Toye, A.A.; Dumas, M.E.; Blancher, C.; Rothwell, A.R.; Fearnside, J.F.; Wilder, S.P.; Bihoreau, M.T.; Cloarec, O.; Azzouzi, I.; Young, S.; et al. Subtle metabolic and liver gene transcriptional changes underlie diet-induced fatty liver susceptibility in insulin-resistant mice. Diabetologia 2007, 50, 1867–1879. [Google Scholar] [CrossRef]
- Walle, P.; Takkunen, M.; Mannisto, V.; Vaittinen, M.; Lankinen, M.; Karja, V.; Kakela, P.; Agren, J.; Tiainen, M.; Schwab, U.; et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism 2016, 65, 655–666. [Google Scholar] [CrossRef]
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017, 37 (Suppl. S1), 97–103. [Google Scholar] [CrossRef]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Siren, J.; Hamsten, A.; Fisher, R.M.; Yki-Jarvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, G.S.; DiStefano, J.K. Micro RNAs in the development of non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 226–234. [Google Scholar] [CrossRef]
- Powell, C.A.; Choudhury, M. Advancing metabolism research to overcome low litter survival in metabolically stressed mice. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E261–E268. [Google Scholar] [CrossRef] [PubMed]
- Meruvu, S.; Zhang, J.; Choudhury, M. Mono-(2-ethylhexyl) Phthalate Increases Oxidative Stress Responsive miRNAs in First Trimester Placental Cell Line HTR8/SVneo. Chem. Res. Toxicol. 2016, 29, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Choudhury, M. The plasticizer BBP selectively inhibits epigenetic regulator sirtuin during differentiation of C3H10T1/2 stem cell line. Toxicol. In Vitro 2017, 39, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Meruvu, S.; Zhang, J.; Bedi, Y.S.; Choudhury, M. Mono-(2-ethylhexyl) phthalate induces apoptosis through miR-16 in human first trimester placental cell line HTR-8/SVneo. Toxicol. In Vitro 2016, 31, 35–42. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Powell, C.A.; Kay, M.K.; Sonkar, R.; Meruvu, S.; Choudhury, M. Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. Int. J. Mol. Sci. 2021, 22, 1822. https://doi.org/10.3390/ijms22041822
Zhang J, Powell CA, Kay MK, Sonkar R, Meruvu S, Choudhury M. Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. International Journal of Molecular Sciences. 2021; 22(4):1822. https://doi.org/10.3390/ijms22041822
Chicago/Turabian StyleZhang, Jian, Catherine A Powell, Matthew K Kay, Ravi Sonkar, Sunitha Meruvu, and Mahua Choudhury. 2021. "Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation" International Journal of Molecular Sciences 22, no. 4: 1822. https://doi.org/10.3390/ijms22041822
APA StyleZhang, J., Powell, C. A., Kay, M. K., Sonkar, R., Meruvu, S., & Choudhury, M. (2021). Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. International Journal of Molecular Sciences, 22(4), 1822. https://doi.org/10.3390/ijms22041822