
Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation

 ,
, 

Abstract
1. Introduction
2. Results
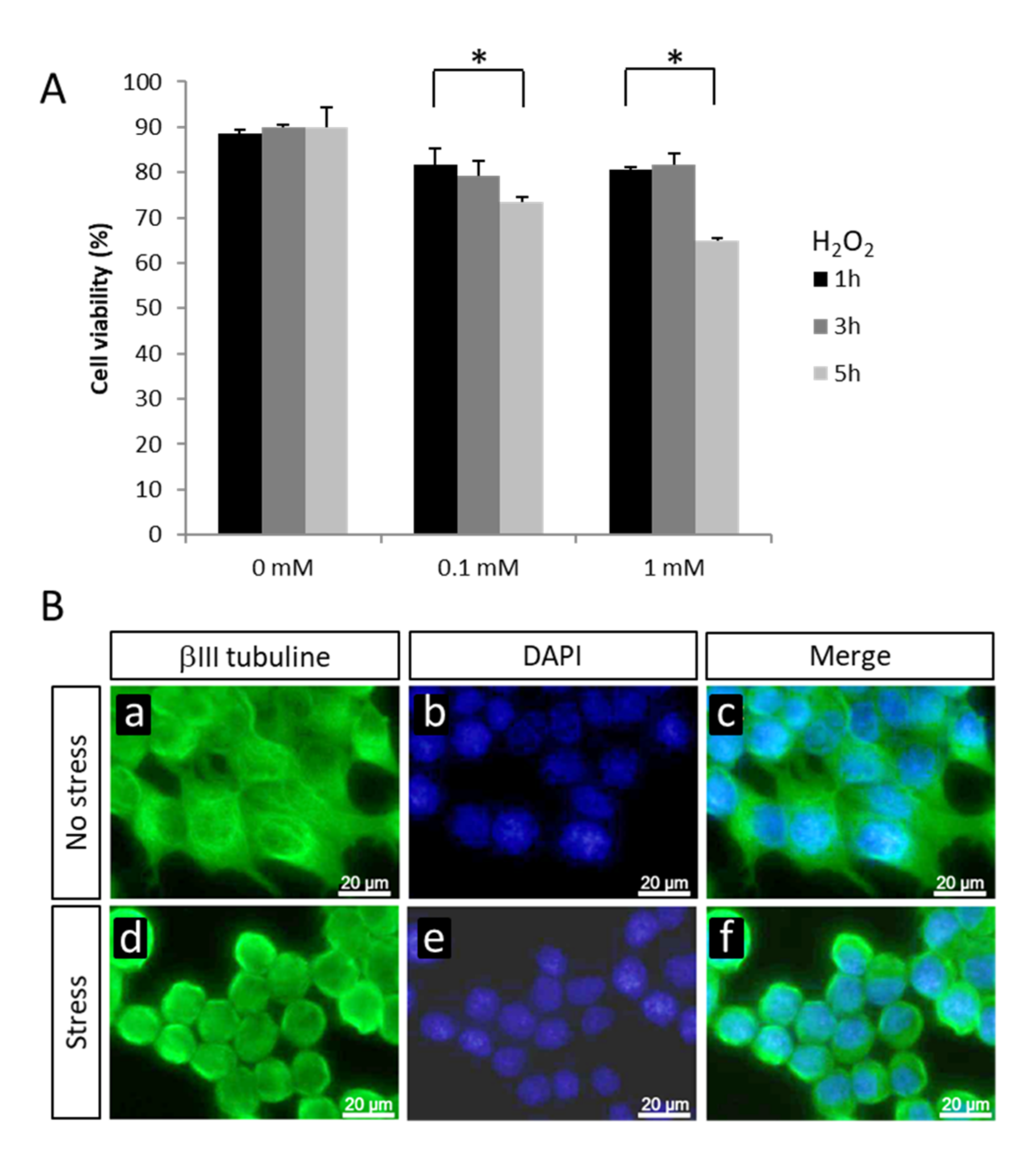
2.1. Changes in Gene Expression in NSC-34 Cells Exposed to Oxidative Stress
2.2. Gene Expression Variations in the Ubiquitin/Ubiquitin-like Pathways
2.3. Combined Effect of Oxidative Stress and Mutant SOD1A4V
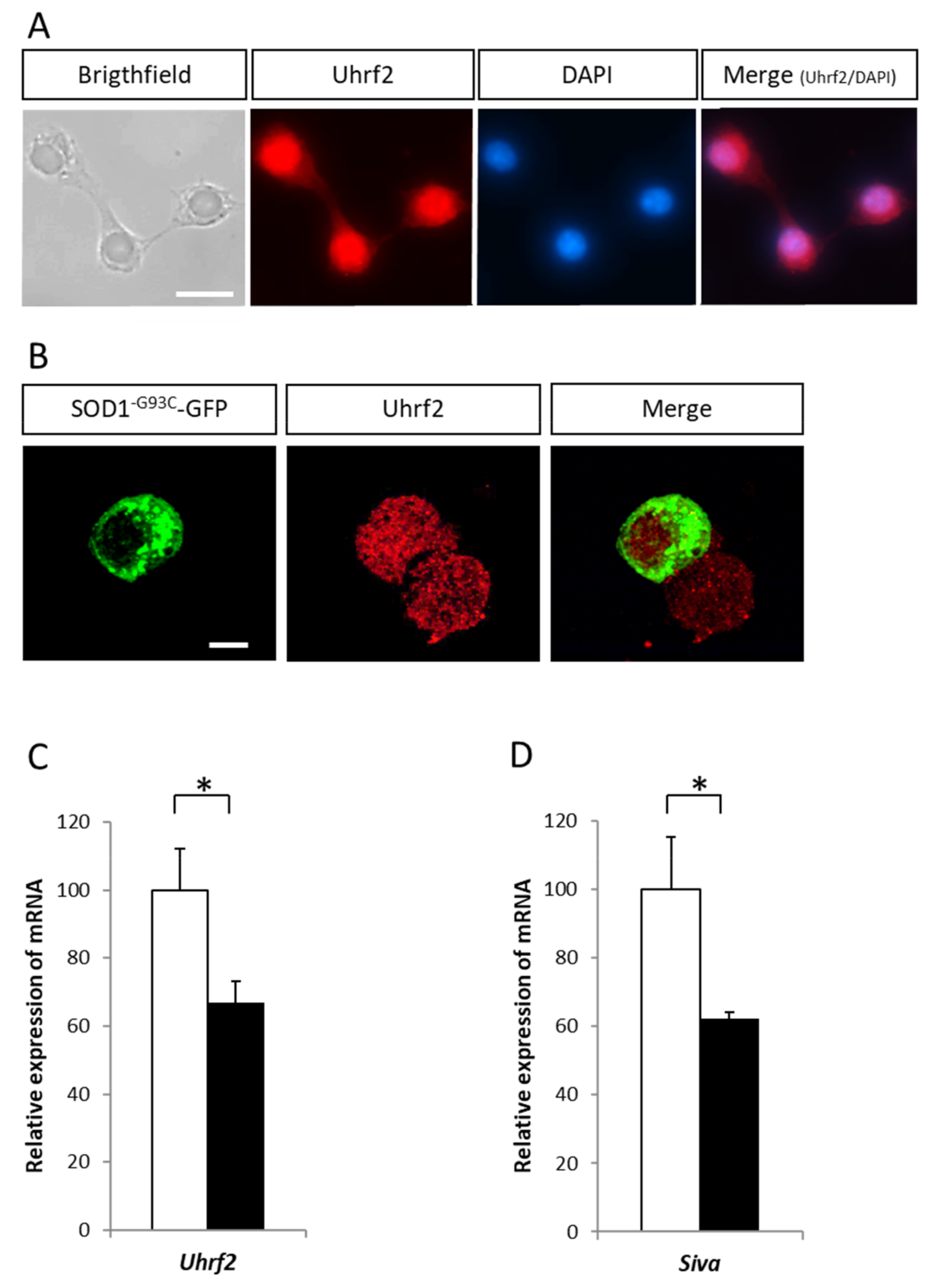
2.4. Variation of Expression of the E3 Ligase Uhrf2 in Presence of SOD1 Mutants
3. Discussion
4. Methods
4.1. Cell Cultures
4.2. Cell Viability Assay
4.3. Microarray Experiments
4.4. Expression Vectors and Transfections
4.5. Western Blot Analysis
4.6. Immunocytochemical Analysis
4.7. RT-qPCR Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Dangoumau, A.; Veyrat-Durebex, C.; Blasco, H.; Praline, J.; Corcia, P.; Andres, C.R.; Vourc’H, P. Protein SUMOylation, an emerging pathway in amyotrophic lateral sclerosis. Int. J. Neurosci. 2013, 123, 366–374. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’H, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L.; Isaacs, A.M. Ageing as a risk factor for ALS/FTD. Hum. Mol. Genet. 2017, 26, R105–R113. [Google Scholar] [CrossRef]
- Kuraszkiewicz, B.; Podsiadły-Marczykowska, T.; Goszczyńska, H.; Piotrkiewicz, M. Are There Modifiable Environmental Factors Related to Amyotrophic Lateral Sclerosis? Front. Neurol. 2018, 9, 220. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell Longev. 2020, 2020, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 69, 2064–2074. [Google Scholar] [CrossRef]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Camu, W.; Brulard, C.; Marouillat, S.; Couratier, P.; Camdessanche, J.P.; Cintas, P.; Vershueren, A.; Soriani, M.H.; Desnuelle, C.; et al. Effect of Familial clustering in the genetic screening in 235 French ALS families. J. Neurol. Neurosurg. Psychiatry 2021. [Google Scholar] [CrossRef]
- Eleutherio, E.C.A.; Silva Magalhães, R.S.; de Araújo Brasil, A.; Monteiro Neto, J.R.; de Holanda Paranhos, L. SOD1, More than just an antioxidant. Arch. Biochem. Biophys. 2020, 697, 108701. [Google Scholar] [CrossRef] [PubMed]
- Guégan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the Mitochondrial-Dependent Apoptotic Pathway in Amyotrophic Lateral Sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Althaus, J.S.; Ellerbrock, B.R.; Becker, D.A.; Gurney, M.E. Enhanced oxygen radical production in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1998, 44, 763–770. [Google Scholar] [CrossRef]
- Watanabe, M.; Dykes-Hoberg, M.; Culotta, V.C.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological Evidence of Protein Aggregation in Mutant SOD1 Transgenic Mice and in Amyotrophic Lateral Sclerosis Neural Tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Jeon, G.S.; Sung, J. ALS-Linked Mutant SOD1 Associates with TIA-1 and Alters Stress Granule Dynamics. Neurochem. Res. 2020, 45, 2884–2893. [Google Scholar] [CrossRef]
- Hozumi, I. Roles and therapeutic potential of metallothioneins in neurodegenerative diseases. Curr. Pharm. Biotechnol. 2013, 14, 408–413. [Google Scholar] [CrossRef]
- Ono, S.-I. Metallothionein is a Potential Therapeutic Strategy for Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2018, 23, 5001–5009. [Google Scholar] [CrossRef]
- Michelle, C.; Vourc’H, P.; Mignon, L.; Andres, C.R. What Was the Set of Ubiquitin and Ubiquitin-Like Conjugating Enzymes in the Eukaryote Common Ancestor? J. Mol. Evol. 2009, 68, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Topp, S.D.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef]
- Perrelet, D.; Perrin, F.E.; Liston, P.; Korneluk, R.G.; MacKenzie, A.; Ferrer-Alcón, M.; Kato, A.C. Motoneuron Resistance to Apoptotic Cell Death In Vivo Correlates with the Ratio between X-Linked Inhibitor of Apoptosis Proteins (XIAPs) and Its Inhibitor, XIAP-Associated Factor 1. J. Neurosci. 2004, 24, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Nagashima, Y.; Matsumoto, L.; Suzuki, T.; Yamanaka, T.; Date, H.; Deoka, K.; Nukina, N.; Tsuji, S. Intranuclear Degradation of Polyglutamine Aggregates by the Ubiquitin-Proteasome System. J. Biol. Chem. 2009, 284, 9796–9803. [Google Scholar] [CrossRef]
- Mori, T.; Ikeda, D.D.; Yamaguchi, Y.; Unoki, M. NIRF/UHRF2 occupies a central position in the cell cycle network and allows coupling with the epigenetic landscape. FEBS Lett. 2012, 586, 1570–1583. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic Correction of a LRRK2 Mutation in Human iPSCs Links Parkinsonian Neurodegeneration to ERK-Dependent Changes in Gene Expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef]
- Lu, H.; Hallstrom, T.C. The Nuclear Protein UHRF2 Is a Direct Target of the Transcription Factor E2F1 in the Induction of Apoptosis. J. Biol. Chem. 2013, 288, 23833–23843. [Google Scholar] [CrossRef] [PubMed]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Durham, H.D.; Dahrouge, S.; Cashman, N.R. Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. NeuroToxicology 1993, 14, 387–395. [Google Scholar]
- Maier, O.; Böhm, J.; Dahm, M.; Brück, S.; Beyer, C.; Johann, S. Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem. Int. 2013, 62, 1029–1038. [Google Scholar] [CrossRef]
- Heck, M.V.; Azizov, M.; Stehning, T.; Walter, M.; Kedersha, N.; Auburger, G. Dysregulated expression of lipid storage and membrane dynamics factors in Tia1 knockout mouse nervous tissue. Neurogenetics 2014, 15, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cook, A.; Kim, J.; Baranov, S.V.; Jiang, J.; Smith, K.; Cormier, K.; Bennett, E.; Browser, R.P.; Day, A.L.; et al. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 55, 26–35. [Google Scholar] [CrossRef]
- Tokuda, E.; Okawa, E.; Watanabe, S.; Ono, S. Overexpression of metallothionein-I, a copper-regulating protein, attenuates intracellular copper dyshomeostasis and extends lifespan in a mouse model of amyotrophic lateral sclerosis caused by mutant superoxide dismutase-1. Hum. Mol. Genet. 2014, 23, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Kamitani, T.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson’s disease and dementia with Lewy bodies. Neurobiol. Dis. 2010, 38, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wang, Y.; Luo, Z.; Chang, L.-C.; Yoo, J.S.; Yan, H.; Choi, Y.; Xie, X.; Deverman, B.E.; Gradinaru, V.; et al. TRIM9-Mediated Resolution of Neuroinflammation Confers Neuroprotection upon Ischemic Stroke in Mice. Cell Rep. 2019, 27, 549–560.e6. [Google Scholar] [CrossRef]
- Winkle, C.C.; McClain, L.M.; Valtschanoff, J.G.; Park, C.S.; Maglione, C.; Gupton, S.L. A novel Netrin-1–sensitive mechanism promotes local SNARE-mediated exocytosis during axon branching. J. Cell Biol. 2014, 205, 217–232. [Google Scholar] [CrossRef]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free. Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Kanno, T.; Tanaka, K.; Yanagisawa, Y.; Yasutake, K.; Hadano, S.; Yoshii, F.; Hirayama, N.; Ikeda, J.-E. A novel small molecule, N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide, selectively protects against oxidative stress-induced cell death by activating the Nrf2–ARE pathway: Therapeutic implications for ALS. Free. Radic. Biol. Med. 2012, 53, 2028–2042. [Google Scholar] [CrossRef]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Aerobic glycolysis in amyotrophic lateral sclerosis and Huntington’s disease. Rev. Neurosci. 2018, 29, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Cárdenas, P.; Osses, N.; Henríquez, J.P. Characterization of Wnt/β-catenin and BMP/Smad signaling pathways in an in vitro model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2013, 7, 239. [Google Scholar] [CrossRef]
- Yu, Y.; Nakagawa, T.; Morohoshi, A.; Nakagawa, M.; Ishida, N.; Suzuki, N.; Aoki, M.; Nakayama, K. Pathogenic mutations in the ALS gene CCNF cause cytoplasmic mislocalization of Cyclin F and elevated VCP ATPase activity. Hum. Mol. Genet. 2019, 28, 3486–3497. [Google Scholar] [CrossRef] [PubMed]
- Dangoumau, A.; Verschueren, A.; Hammouche, E.; Papon, M.-A.; Blasco, H.; Cherpi-Antar, C.; Pouget, J.; Corcia, P.; Andres, C.R.; Vourc’H, P. A novel SOD1 mutation p.V31A identified with a slowly progressive form of amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 266.e1–266.e4. [Google Scholar] [CrossRef]
- Régal, L.; Vanopdenbosch, L.; Tilkin, P.; Van den Bosch, L.; Thijs, V.; Sciot, R.; Robberecht, W. The G93C mutation in superoxide dismutase 1: Clinicopathologic phenotype and prognosis. Arch. Neurol. 2006, 63, 262–267. [Google Scholar] [CrossRef]
- Saeed, M.; Yang, Y.; Deng, H.-X.; Hung, W.-Y.; Siddique, T.; Dellefave, L.; Gellera, C.; Andersen, P.M. Age and founder effect of SOD1 A4V mutation causing ALS. Neurology 2009, 72, 1634–1639. [Google Scholar] [CrossRef]
- Cookson, M.R.; Menzies, F.M.; Manning, P.; Eggett, C.J.; Figlewicz, D.A.; McNeil, C.J.; Shaw, P.J. Cu/Zn superoxide dismutase (SOD1) mutations associated with familial amyotrophic lateral sclerosis (ALS) affect cellular free radical release in the presence of oxidative stress. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2002, 3, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. p53 Is Abnormally Elevated and Active in the CNS of Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef]
- Peuget, S.; Bonacci, T.; Soubeyran, P.; Iovanna, J.L.; Dusetti, N. Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death Differ. 2014, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Saville, M.K.; Sparks, A.; Xirodimas, D.P.; Wardrop, J.; Stevenson, L.F.; Bourdon, J.-C.; Woods, Y.L.; Lane, D.P. Regulation of p53 by the ubiquitin-conjugating enzymes UbcH5B/C in vivo. J. Biol. Chem. 2004, 279, 42169–42181. [Google Scholar] [CrossRef]
- Boutahar, N.; Wierinckx, A.; Camdessanche, J.P.; Antoine, J.-C.; Reynaud, E.; Lassabliere, F.; Lachuer, J.; Borg, J. Differential effect of oxidative or excitotoxic stress on the transcriptional profile of amyotrophic lateral sclerosis-linked mutant SOD1 cultured neurons. J. Neurosci. Res. 2011, 89, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.P.; Alves, C.J.; Chadi, G. Early gene expression changes in spinal cord from SOD1G93A Amyotrophic Lateral Sclerosis animal model. Front. Cell. Neurosci. 2013, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Smith, J.A.; Gibson, C.; Varma, A.K.; Ray, S.K.; Banik, N.L. Estrogen receptor agonists and estrogen attenuate TNF-α-induced apoptosis in VSC4.1 motoneurons. J. Endocrinol. 2010, 208, 171–182. [Google Scholar] [CrossRef]
- Oh, Y.; Chung, K.C. UHRF2, a Ubiquitin E3 Ligase, Acts as a Small Ubiquitin-like Modifier E3 Ligase for Zinc Finger Protein 131*. J. Biol. Chem. 2013, 288, 9102–9111. [Google Scholar] [CrossRef]
- Trappe, R.; Buddenberg, P.; Uedelhoven, J.; Gläser, B.; Buck, A.; Engel, W.; Burfeind, P. The murine BTB/POZ zinc finger gene Znf131: Predominant expression in the developing central nervous system, in adult brain, testis, and thymus. Biochem. Biophys. Res. Commun. 2002, 296, 319–327. [Google Scholar] [CrossRef]
- Jacobs, S.B.R.; Basak, S.; I Murray, J.; Pathak, N.; Attardi, L.D. Siva is an apoptosis-selective p53 target gene important for neuronal cell death. Cell Death Differ. 2007, 14, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Broom, W.J.; Russ, C.; Sapp, P.C.; McKenna-Yasek, D.; Hosler, B.A.; Andersen, P.M.; Brown, R.H. Variants in candidate ALS modifier genes linked to Cu/Zn superoxide dismutase do not explain divergent survival phenotypes. Neurosci. Lett. 2006, 392, 52–57. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Fold Change | ||
|---|---|---|---|---|
| Cellular response to DNA damage stimulus | ||||
| Cep164 | centrosomal protein 164 | −1.80 | ||
| E2f7 | E2F transcription factor 7 | −1.35 | ||
| Ube2b | Ubiquitin-conjugating enzyme E2B | 1.31 | ||
| Rbx1 | ring-box 1 | 1.35 | ||
| Brip1 | BRCA1 interacting protein C-terminal helicase 1 | 1.36 | ||
| Pcna | proliferating cell nuclear antigen | 1.37 | ||
| Fancl | Fanconi anemia, complementation group L | 1.47 | ||
| Gtf2h1 | general transcription factor II H, polypeptide 1 | 1.56 | ||
| Apoptotic process | ||||
| Myc | myelocytomatosis oncogene | −2.83 | ||
| Tia1 | cytotoxic granule-associated RNA binding protein 1 | −2.53 | ||
| Pde5a | phosphodiesterase 5A, cGMP-specific | −2.21 | ||
| Mkl1 | MKL (megakaryoblastic leukemia)/myocardin-like 1 | −1.82 | ||
| Bik | BCL2-interacting killer | −1.71 | ||
| Dusp1 | dual specificity phosphatase 1 | −1.65 | ||
| Pim1 | proviral integration site 1 | −1.46 | ||
| Senp1 | SUMO1/sentrin specific peptidase 1 | −1.34 | ||
| Ube2b | Ubiquitin-conjugating enzyme E2B | 1.31 | ||
| Tm2d1 | TM2 domain containing 1 | 1.38 | ||
| Birc5 | baculoviral IAP repeat-containing 5 | 1.41 | ||
| Mad2l1 | MAD2 mitotic arrest deficient-like 1 | 1.43 | ||
| Cfdp1 | craniofacial development protein 1 | 1.56 | ||
| Cycs | cytochrome c, somatic | 1.58 | ||
| Mt1 | metallothionein 1 | 7.51 | ||
| Antioxidant response | ||||
| Pdia2 | protein disulfide isomerase associated 2 | −2.71 | ||
| Mtf1 | metal response element binding transcription factor 1 | −1.63 | ||
| Synaptic functions | ||||
| Sipa1l1 | signal-induced proliferation-associated 1 like 1 | −1.81 | ||
| Btbd9 | BTB (POZ) domain containing 9 | −1.79 | ||
| Apba1 | amyloid beta (A4) precursor protein binding. family A, member 1 | −1.79 | ||
| Nat8l | N-acetyltransferase 8-like | −1.65 | ||
| Pacsin1 | protein kinase C and casein kinase substrate in neurons 1 | −1.34 | ||
| Paip2 | polyadenylate-binding protein-interacting protein 2 | 1.55 | ||
| Cnn3 | calponin 3, acidic | 2.25 | ||
| Mitochondrial function | ||||
| Bik | BCL2-interacting killer | −1.71 | ||
| Alas1 | aminolevulinic acid synthase 1 | −1.43 | ||
| Slmo2 | slowmo homolog 2 (Drosophila) | 1.32 | ||
| Trit1 | tRNA isopentenyltransferase 1 | 1.35 | ||
| Metap1d | methionyl aminopeptidase type 1D (mitochondrial) | 1.36 | ||
| Lypla1 | lysophospholipase 1 | 1.38 | ||
| Hibadh | 3-hydroxyisobutyrate dehydrogenase | 1.38 | ||
| Mrpl9 | mitochondrial ribosomal protein L9 | 1.41 | ||
| Pgam5 | phosphoglycerate mutase family member 5 | 1.42 | ||
| Timm8a1 | translocase of inner mitochondrial membrane 8A1 | 1.49 | ||
| Ndufab1 | NADH dehydrogenase (ubiquinone) 1, alpha/beta subcomplex, 1 | 1.54 | ||
| Lyrm7 | LYR motif containing 7 | 2.03 | ||
| Endosome and golgi functions | ||||
| Adcy6 | adenylate cyclase 6 | −1.53 | ||
| Rab21 | RAB21, member RAS oncogene family | −1.35 | ||
| Rab9 | RAB9, member RAS oncogene family | 1.33 | ||
| Pmel | premelanosome protein | 1.50 | ||
| Arl1 | ADP-ribosylation factor-like 1 | 1.52 | ||
| Gene Symbol | Gene Name | Fold Change |
|---|---|---|
| E2 conjugating enzymes | ||
| Ube2d1 | Ubiquitin-conjugating enzyme E2D1 | −1.87 |
| Ube2d2 | Ubiquitin-conjugating enzyme E2D2 | 1.30 |
| Ube2b | Ubiquitin-conjugating enzyme E2B | 1.31 |
| Ube2e1 | Ubiquitin-conjugating enzyme E2E1 | 1.37 |
| Ube2c | Ubiquitin-conjugating enzyme E2C | 1.39 |
| E3 ligases | ||
| E3 ligases with multipled subunits with RING domain | ||
| Subunits with RING domain | ||
| Rbx1 | Ring-box 1 | 1.35 |
| Subunits (adaptators) | ||
| CCNF | Cyclin F baisse | −2.44 |
| Spsb4 | SpIA/ryanodine receptor domain and SOCS box containing 4 | −1.92 |
| Btbd9 | BTB (POZ) domain containing 9 | −1.78 |
| Klhl29 | Kelch-like 29 (Drosophila) | −1.61 |
| Fbxo46 | F-box protein 46 | −1.54 |
| Rhobtb2 | Rho-related BTB domain containing 2 | −1.43 |
| Klhl21 | Kelch-like 21 (Drosophila) | −1.33 |
| Fbxo42 | F-box protein 42 | −1.26 |
| Cdc16 | CDC16 cell division cycle 16 homolog (S.cerevisiae) | 1.30 |
| Zbtb32 | Zinc finger and BTB domain containing 32 | 1.41 |
| other E3 ligases | ||
| Trim9 | Tripartite motif-containing 9, transcript variant 3 | −2.27 |
| Sh3rf1 | SH3 domain containing RING finger 1 | −1.95 |
| Rnf121 | RING finger protein 121 | 1.35 |
| Fancl | Fanconi anemia, complementation group L | 1.46 |
| Uhrf2 | Ubiquitin-like, containing PHD and RING finger domains 2 | 1.49 |
| Others | ||
| Usp44 | Ubiquitin specific peptidase 44 | −2.25 |
| Usp36 | Ubiquitin specific peptidase 36 | −1.70 |
| Senp1 | SUMO1/sentrin specific peptidase 1 | −1.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dangoumau, A.; Marouillat, S.; Coelho, R.; Wurmser, F.; Brulard, C.; Haouari, S.; Laumonnier, F.; Corcia, P.; Andres, C.R.; Blasco, H.; et al. Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation. Int. J. Mol. Sci. 2021, 22, 1796. https://doi.org/10.3390/ijms22041796
Dangoumau A, Marouillat S, Coelho R, Wurmser F, Brulard C, Haouari S, Laumonnier F, Corcia P, Andres CR, Blasco H, et al. Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation. International Journal of Molecular Sciences. 2021; 22(4):1796. https://doi.org/10.3390/ijms22041796
Chicago/Turabian StyleDangoumau, Audrey, Sylviane Marouillat, Roxane Coelho, François Wurmser, Céline Brulard, Shanez Haouari, Frédéric Laumonnier, Philippe Corcia, Christian R. Andres, Hélène Blasco, and et al. 2021. "Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation" International Journal of Molecular Sciences 22, no. 4: 1796. https://doi.org/10.3390/ijms22041796
APA StyleDangoumau, A., Marouillat, S., Coelho, R., Wurmser, F., Brulard, C., Haouari, S., Laumonnier, F., Corcia, P., Andres, C. R., Blasco, H., & Vourc’h, P. (2021). Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation. International Journal of Molecular Sciences, 22(4), 1796. https://doi.org/10.3390/ijms22041796