
Defective Oligodendroglial Lineage and Demyelination in Amyotrophic Lateral Sclerosis


Abstract
1. Introduction
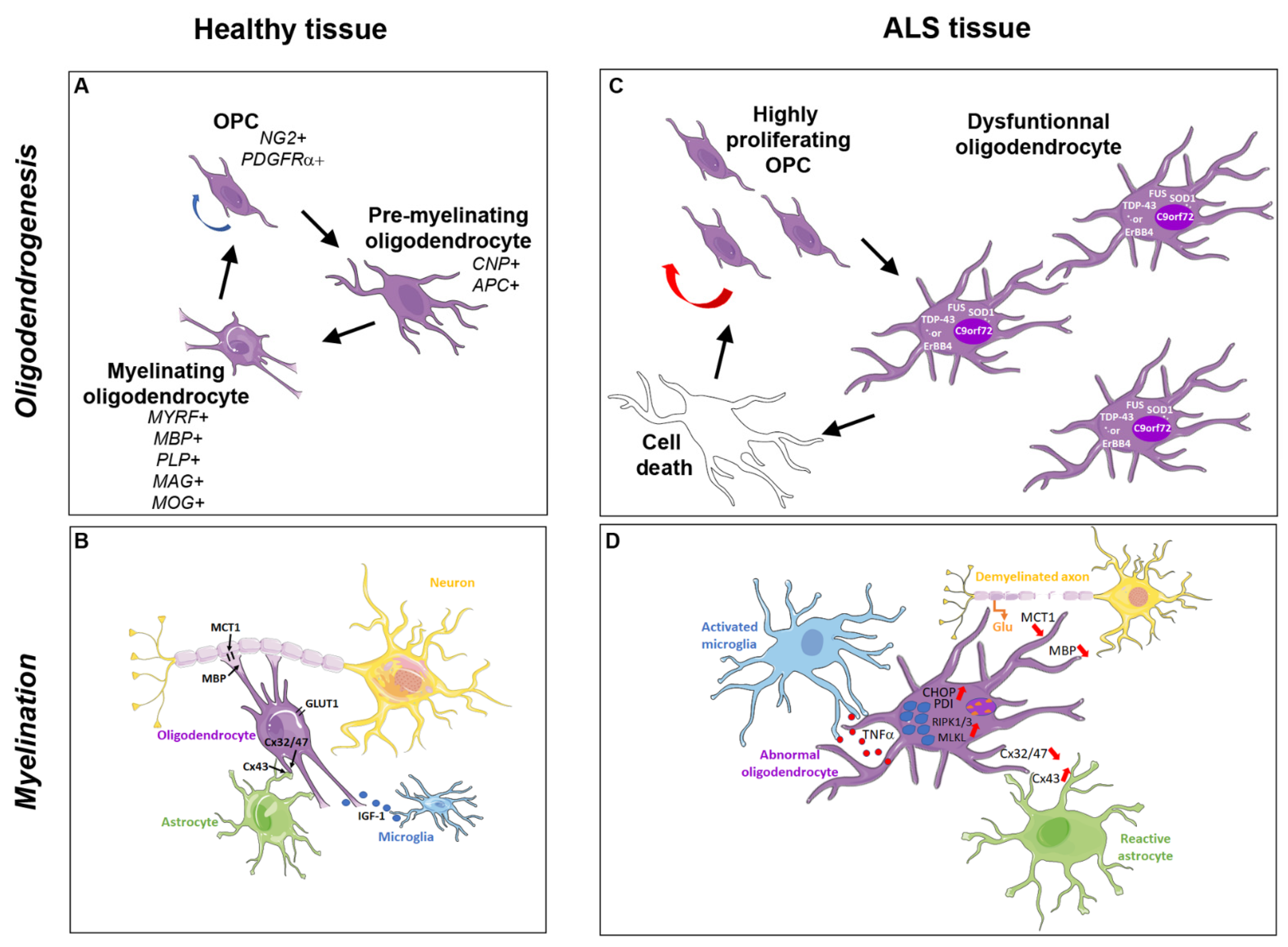
2. The Oligodendroglial Cells and Myelination Process
2.1. Oligodendrocyte Generation
2.2. Axonal Myelination
2.3. Oligodendroglial Metabolic Support to the Axon
3. Pathological Inclusions in Oligodendrocytes from ALS Patients and Animal Models
3.1. TDP-43 Inclusions
3.2. FUS Inclusions
3.3. SOD1 Inclusions
3.4. ErBB4 Inclusions
4. Degenerating and Dysfunctional Oligodendrocytes in ALS Patients and Animal Models
4.1. Reduction of MCT1 Expression
4.2. Degeneration and Accelerated Turnover of Oligodendroglial Cells
4.3. Regeneration of Dysfunctional Oligodendrocytes
4.4. Abnormal Myelination
4.5. Motor Neuron Death Induced by ALS Gene Expression in Oligodendrocytes
5. Molecular Mechanisms Involved in Oligodendrocyte Dysfunction and Degeneration in ALS
5.1. Glutamate Excitotoxicity Induced by Motor Neuron Death
5.2. Abnormalities in Protein Homeostasis
5.3. Oxidative Damage to mRNAs Involved in Myelination
5.4. Deleterious Effects Involving Astrocytes
5.5. Deleterious Effects Involving Microglia
6. Oligodendrocyte Degeneration as a Primary or Secondary Event in ALS Pathogenesis
6.1. Oligodendrocyte Degeneration as a Primary Event
6.2. Oligodendrocyte Degeneration as a Secondary Event
6.3. Primary or Secondary Event: A Yet Unanswered Question
6.4. The Hypothesis of the Putative Aberrant Regulation of Olig2 Function
7. Therapeutic Perspectives Provided by Oligodendrocyte Implication in the Pathogenesis of ALS
7.1. Available Treatments and Antisense Oligonucleotides
7.2. Oligodendrocytes as Targets for Future Treatments in ALS

{kind=link}
| Oligodendrocyte Defect. | Potential Therapy | ALS Patients | Animal Models |
|---|---|---|---|
| TDP-43 inclusions | -ASO for Ataxin-2 | TDP-43 Tg: extends lifespan and reduces pathology [156] | |
| SOD1 inclusions | -ASO | -Phase 1–2: decreases SOD1 concentration in CSF [158] | |
| RNA foci C9orf72 | -ASO | C9orf72 Tg: protects against ALS [155] | |
| ErBB4 inclusions | -ASO | - | - |
| Myelin degeneration | Promyelinating molecules -Clemastine (Anti-Histamine H1) -Montelukast (Anti-GPR17) -Bexarotene (RXR-γ agonist) -Tamoxifen | -Mitigates progression [159] | -Prolongs survival in SODG93A [160] -Rescues SODG93A OPC differentiation in vitro [161] -Prolongs survival in SODG93A [162] |
| Altered trophic support to the axon | Rescue of nutrient flow -Co-factor of carboxylases to produce ATP (Biotin) -Mitochondrial ATP synthesis (RNS60) | -Pilot study: safe and well tolerated [163] -Pilot study: safe and well tolerated [164] | -Prolongs survival in SODG93A [165] |
| OL degeneration via: -Glutamate excitotoxicity -Oxidative stress -Neuroinflammation | OL replacement -Anti-glutamatergic (Riluzole) -Anti-oxidative agents (Edavarone) -Modulators of reactive gliosis (Masitinib) (Clemastine) (Cannabinoid receptor 2 antagonist) | -Several Phases 1: delayed disease progression -Approved -Approved -Phase 3 [148,149] | -Prolongs survival in SODG93A [160] -Limits progression in TDP-43 Tg [166] |
7.2.1. Therapeutic Approaches Requiring Improvements
7.2.2. Promyelinating Molecules
8. Conclusions
Funding
Conflicts of Interest
References
- Morris, J. Amyotrophic Lateral Sclerosis (ALS) and Related Motor Neuron Diseases: An Overview. Neurodiagn. J. 2015, 55, 180–194. [Google Scholar] [CrossRef]
- Marin, B.; Boumediene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Dion, P.A.; Rouleau, G.A. ALS: Recent Developments from Genetics Studies. Curr. Neurol. Neurosci. Rep. 2016, 16, 59. [Google Scholar] [CrossRef]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Pape, J.A.; Grose, J.H. The effects of diet and sex in amyotrophic lateral sclerosis. Rev. Neurol. 2020, 176, 301–315. [Google Scholar] [CrossRef]
- Bozzoni, V.; Pansarasa, O.; Diamanti, L.; Nosari, G.; Cereda, C.; Ceroni, M. Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 2016, 31, 7–19. [Google Scholar] [CrossRef]
- Chio, A.; Mora, G.; Moglia, C.; Manera, U.; Canosa, A.; Cammarosano, S.; Ilardi, A.; Bertuzzo, D.; Bersano, E.; Cugnasco, P.; et al. Secular Trends of Amyotrophic Lateral Sclerosis: The Piemonte and Valle d’Aosta Register. JAMA Neurol. 2017, 74, 1097–1104. [Google Scholar] [CrossRef]
- Curzio, D.D.; Gurm, M.; Turnbull, M.; Nadeau, M.J.; Meek, B.; Rempel, J.D.; Fineblit, S.; Jonasson, M.; Hebert, S.; Ferguson-Parry, J.; et al. Pro-Inflammatory Signaling Upregulates a Neurotoxic Conotoxin-Like Protein Encrypted Within Human Endogenous Retrovirus-K. Cells 2020, 9, 1584. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Mathis, S.; Couratier, P.; Julian, A.; Vallat, J.M.; Corcia, P.; Le Masson, G. Management and therapeutic perspectives in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2017, 17, 263–276. [Google Scholar] [CrossRef]
- Vucic, S.; Lin, C.S.; Cheah, B.C.; Murray, J.; Menon, P.; Krishnan, A.V.; Kiernan, M.C. Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 2013, 136, 1361–1370. [Google Scholar] [CrossRef]
- Bissaro, M.; Moro, S. Rethinking to riluzole mechanism of action: The molecular link among protein kinase CK1delta activity, TDP-43 phosphorylation, and amyotrophic lateral sclerosis pharmacological treatment. Neural Regen Res. 2019, 14, 2083–2085. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler 2006, 7, 241–245. [Google Scholar] [CrossRef]
- Wootz, H.; Fitzsimons-Kantamneni, E.; Larhammar, M.; Rotterman, T.M.; Enjin, A.; Patra, K.; Andre, E.; Van Zundert, B.; Kullander, K.; Alvarez, F.J. Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J. Comp. Neurol. 2013, 521, 1449–1469. [Google Scholar] [CrossRef]
- Ng, A.S.; Rademakers, R.; Miller, B.L. Frontotemporal dementia: A bridge between dementia and neuromuscular disease. Ann. N. Y. Acad. Sci. 2015, 1338, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hughes, E.G.; Shetty, A.S.; Arlotta, P.; Goff, L.A.; Bergles, D.E.; Brown, S.P. Changes in the Excitability of Neocortical Neurons in a Mouse Model of Amyotrophic Lateral Sclerosis Are Not Specific to Corticospinal Neurons and Are Modulated by Advancing Disease. J. Neurosci. 2017, 37, 9037–9053. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.K.; Sutherland, N.M.; Zhang, S.; Hatzipetros, T.; Vieira, F.; Valdez, G. The ALS-inducing factors, TDP43(A315T) and SOD1(G93A), directly affect and sensitize sensory neurons to stress. Sci. Rep. 2018, 8, 16582. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Boillee, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Rafalowska, J.; Dziewulska, D. White matter injury in amyotrophic lateral sclerosis (ALS). Folia Neuropathol. 1996, 34, 87–91. [Google Scholar] [PubMed]
- Sach, M.; Winkler, G.; Glauche, V.; Liepert, J.; Heimbach, B.; Koch, M.A.; Buchel, C.; Weiller, C. Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain 2004, 127, 340–350. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Ansorge, O.; Strong, M.; Bilbao, J.; Zinman, L.; Ang, L.C.; Baker, M.; Stewart, H.; Eisen, A.; Rademakers, R.; et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011, 122, 87–98. [Google Scholar] [CrossRef]
- Neumann, M.; Kwong, L.K.; Truax, A.C.; Vanmassenhove, B.; Kretzschmar, H.A.; Van Deerlin, V.M.; Clark, C.M.; Grossman, M.; Miller, B.L.; Trojanowski, J.Q.; et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 2007, 66, 177–183. [Google Scholar] [CrossRef]
- Niebroj-Dobosz, I.; Rafalowska, J.; Fidzianska, A.; Gadamski, R.; Grieb, P. Myelin composition of spinal cord in a model of amyotrophic lateral sclerosis (ALS) in SOD1G93A transgenic rats. Folia Neuropathol. 2007, 45, 236–241. [Google Scholar]
- Seilhean, D.; Cazeneuve, C.; Thuries, V.; Russaouen, O.; Millecamps, S.; Salachas, F.; Meininger, V.; Leguern, E.; Duyckaerts, C. Accumulation of TDP-43 and alpha-actin in an amyotrophic lateral sclerosis patient with the K17I ANG mutation. Acta Neuropathol. 2009, 118, 561–573. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Traiffort, E.; Zakaria, M.; Laouarem, Y.; Ferent, J. Hedgehog: A key signaling in the development of the oligodendrocyte lineage. J. Dev. Biol. 2016, 4, 28. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; McClain, C.R.; Schanz, S.J.; Protack, T.L.; Windrem, M.S.; Goldman, S.A. CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nat. Biotechnol. 2011, 29, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I. Oligodendrogliopathy in neurodegenerative diseases with abnormal protein aggregates: The forgotten partner. Prog. Neurobiol. 2018, 169, 24–54. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Richardson, W.D. Evolution of the CNS myelin gene regulatory program. Brain Res. 2016, 1641, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, C.; Bertalot, T.; Zusso, M.; Skaper, S.D.; Giusti, P. Systematic Review of Pharmacological Properties of the Oligodendrocyte Lineage. Front. Cell. Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- Miller, R.H. Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol. 2002, 67, 451–467. [Google Scholar] [CrossRef]
- Nishiyama, A.; Boshans, L.; Goncalves, C.M.; Wegrzyn, J.; Patel, K.D. Lineage, fate, and fate potential of NG2-glia. Brain Res. 2016, 1638, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.H.; Tripathi, R.B.; Richardson, W.D.; Franklin, R.J. Developmental Origin of Oligodendrocyte Lineage Cells Determines Response to Demyelination and Susceptibility to Age-Associated Functional Decline. Cell Rep. 2016. [Google Scholar] [CrossRef]
- Vigano, F.; Schneider, S.; Cimino, M.; Bonfanti, E.; Gelosa, P.; Sironi, L.; Abbracchio, M.P.; Dimou, L. GPR17 expressing NG2-Glia: Oligodendrocyte progenitors serving as a reserve pool after injury. Glia 2016, 64, 287–299. [Google Scholar] [CrossRef]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcao, A.; Xiao, L.; Li, H.; Haring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef]
- Floriddia, E.M.; Lourenco, T.; Zhang, S.; van Bruggen, D.; Hilscher, M.M.; Kukanja, P.; Goncalves Dos Santos, J.P.; Altinkok, M.; Yokota, C.; Llorens-Bobadilla, E.; et al. Distinct oligodendrocyte populations have spatial preference and different responses to spinal cord injury. Nat. Commun. 2020, 11, 5860. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Lang, J.K.; Waldau, B.; Roy, N.S.; Schwartz, T.E.; Pilcher, W.H.; Chandross, K.J.; Natesan, S.; Merrill, J.E.; Goldman, S.A. Complementary patterns of gene expression by human oligodendrocyte progenitors and their environment predict determinants of progenitor maintenance and differentiation. Ann. Neurol. 2006, 59, 763–779. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Snaidero, N.; Pahler, G.; Frey, S.; Sanchez, P.; Zweckstetter, M.; Janshoff, A.; Schneider, A.; Weil, M.T.; Schaap, I.A.; et al. Myelin membrane assembly is driven by a phase transition of myelin basic proteins into a cohesive protein meshwork. PLoS Biol. 2013, 11, e1001577. [Google Scholar] [CrossRef]
- Muller, C.; Bauer, N.M.; Schafer, I.; White, R. Making myelin basic protein -from mRNA transport to localized translation. Front. Cell. Neurosci. 2013, 7, 169. [Google Scholar] [CrossRef]
- Snaidero, N.; Mobius, W.; Czopka, T.; Hekking, L.H.; Mathisen, C.; Verkleij, D.; Goebbels, S.; Edgar, J.; Merkler, D.; Lyons, D.A.; et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014, 156, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Snaidero, N.; Velte, C.; Myllykoski, M.; Raasakka, A.; Ignatev, A.; Werner, H.B.; Erwig, M.S.; Mobius, W.; Kursula, P.; Nave, K.A.; et al. Antagonistic Functions of MBP and CNP Establish Cytosolic Channels in CNS Myelin. Cell Rep. 2017, 18, 314–323. [Google Scholar] [CrossRef]
- Nave, K.A.; Werner, H.B. Myelination of the nervous system: Mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef]
- Tomassy, G.S.; Berger, D.R.; Chen, H.H.; Kasthuri, N.; Hayworth, K.J.; Vercelli, A.; Seung, H.S.; Lichtman, J.W.; Arlotta, P. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 2014, 344, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Dakin, K.A.; Stevens, B.; Lee, P.R.; Kozlov, S.V.; Stewart, C.L.; Fields, R.D. Astrocytes promote myelination in response to electrical impulses. Neuron 2006, 49, 823–832. [Google Scholar] [CrossRef]
- Camargo, N.; Goudriaan, A.; van Deijk, A.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.J.; Woo, D.H.; Lee, P.R.; Pajevic, S.; Bukalo, O.; Huffman, W.C.; Wake, H.; Basser, P.J.; SheikhBahaei, S.; Lazarevic, V.; et al. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 11832–11837. [Google Scholar] [CrossRef] [PubMed]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef] [PubMed]
- Giera, S.; Deng, Y.; Luo, R.; Ackerman, S.D.; Mogha, A.; Monk, K.R.; Ying, Y.; Jeong, S.J.; Makinodan, M.; Bialas, A.R.; et al. The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat. Commun. 2015, 6, 6121. [Google Scholar] [CrossRef]
- Cho, A.N.; Jin, Y.; Kim, S.; Kumar, S.; Shin, H.; Kang, H.C.; Cho, S.W. Aligned Brain Extracellular Matrix Promotes Differentiation and Myelination of Human-Induced Pluripotent Stem Cell-Derived Oligodendrocytes. ACS Appl. Mater. Interfaces 2019, 11, 15344–15353. [Google Scholar] [CrossRef]
- Griffiths, I.; Klugmann, M.; Anderson, T.; Yool, D.; Thomson, C.; Schwab, M.H.; Schneider, A.; Zimmermann, F.; McCulloch, M.; Nadon, N.; et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 1998, 280, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Kassmann, C.M.; Lappe-Siefke, C.; Baes, M.; Brugger, B.; Mildner, A.; Werner, H.B.; Natt, O.; Michaelis, T.; Prinz, M.; Frahm, J.; et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat. Genet. 2007, 39, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Lappe-Siefke, C.; Goebbels, S.; Gravel, M.; Nicksch, E.; Lee, J.; Braun, P.E.; Griffiths, I.R.; Nave, K.A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 2003, 33, 366–374. [Google Scholar] [CrossRef]
- Boullerne, A.I. The history of myelin. Exp. Neurol. 2016, 283, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Hartline, D.K.; Colman, D.R. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr. Biol. 2007, 17, R29–R35. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Mobius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Hildebrand, C.; Remahl, S.; Persson, H.; Bjartmar, C. Myelinated nerve fibres in the CNS. Prog. Neurobiol. 1993, 40, 319–384. [Google Scholar] [CrossRef]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef]
- Yeung, M.S.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.A.; Yong, V.W. Oligodendrocytes and myelin. Neurol. Clin. 1995, 13, 23–49. [Google Scholar] [CrossRef]
- Ludwin, S.K. The pathobiology of the oligodendrocyte. J. Neuropathol. Exp. Neurol. 1997, 56, 111–124. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Nave, K.A. Adapting brain metabolism to myelination and long-range signal transduction. Glia 2014, 62, 1749–1761. [Google Scholar] [CrossRef]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R., 3rd; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Murray, M.E.; DeJesus-Hernandez, M.; Rutherford, N.J.; Baker, M.; Duara, R.; Graff-Radford, N.R.; Wszolek, Z.K.; Ferman, T.J.; Josephs, K.A.; Boylan, K.B.; et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011, 122, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Arai, K.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Lee, E.B.; Kuwabara, S.; Shibuya, K.; Irwin, D.J.; Fang, L.; et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014, 128, 423–437. [Google Scholar] [CrossRef]
- Kolind, S.; Sharma, R.; Knight, S.; Johansen-Berg, H.; Talbot, K.; Turner, M.R. Myelin imaging in amyotrophic and primary lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 562–573. [Google Scholar] [CrossRef]
- Guipponi, M.; Li, Q.X.; Hyde, L.; Beissbarth, T.; Smyth, G.K.; Masters, C.L.; Scott, H.S. SAGE analysis of genes differentially expressed in presymptomatic TgSOD1G93A transgenic mice identified cellular processes involved in early stage of ALS pathology. J. Mol. Neurosci. 2010, 41, 172–182. [Google Scholar] [CrossRef]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)(G93A) ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [PubMed]
- Golubczyk, D.; Malysz-Cymborska, I.; Kalkowski, L.; Janowski, M.; Coates, J.R.; Wojtkiewicz, J.; Maksymowicz, W.; Walczak, P. The Role of Glia in Canine Degenerative Myelopathy: Relevance to Human Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 5740–5748. [Google Scholar] [CrossRef]
- Ditsworth, D.; Maldonado, M.; McAlonis-Downes, M.; Sun, S.; Seelman, A.; Drenner, K.; Arnold, E.; Ling, S.C.; Pizzo, D.; Ravits, J.; et al. Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 907–922. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Kusters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.F.; Eguchi, H.; Tagawa, A.; Onodera, O.; Iwasaki, T.; Tsujino, A.; Nishizawa, M.; Kakita, A.; Takahashi, H. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 2007, 113, 535–542. [Google Scholar] [CrossRef]
- Lu, Y.; Tang, C.; Zhu, L.; Li, J.; Liang, H.; Zhang, J.; Xu, R. The Overexpression of TDP-43 Protein in the Neuron and Oligodendrocyte Cells Causes the Progressive Motor Neuron Degeneration in the SOD1 G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Biol. Sci. 2016, 12, 1140–1149. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Kirby, J.; Highley, R.; Shaw, P.J. The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 2017, 12, e0179375. [Google Scholar] [CrossRef]
- Wang, J.; Ho, W.Y.; Lim, K.; Feng, J.; Tucker-Kellogg, G.; Nave, K.A.; Ling, S.C. Cell-autonomous requirement of TDP-43, an ALS/FTD signature protein, for oligodendrocyte survival and myelination. Proc. Natl. Acad. Sci. USA 2018, 115, E10941–E10950. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Munoz, D.G.; Kusaka, H.; Yokota, O.; Ishihara, K.; Roeber, S.; Kretzschmar, H.A.; Cairns, N.J.; Neumann, M. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011, 121, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Scekic-Zahirovic, J.; Oussini, H.E.; Mersmann, S.; Drenner, K.; Wagner, M.; Sun, Y.; Allmeroth, K.; Dieterle, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 887–906. [Google Scholar] [CrossRef]
- Ho, W.Y.; Chang, J.C.; Tyan, S.H.; Yen, Y.C.; Lim, K.; Tan, B.S.Y.; Ong, J.; Tucker-Kellogg, G.; Wong, P.; Koo, E.; et al. FUS-mediated dysregulation of Sema5a, an autism-related gene, in FUS mice with hippocampus-dependent cognitive deficits. Hum. Mol. Genet 2019, 28, 3777–3791. [Google Scholar] [CrossRef] [PubMed]
- Guzman, K.M.; Brink, L.E.; Rodriguez-Bey, G.; Bodnar, R.J.; Kuang, L.; Xing, B.; Sullivan, M.; Park, H.J.; Koppes, E.; Zhu, H.; et al. Conditional depletion of Fus in oligodendrocytes leads to motor hyperactivity and increased myelin deposition associated with Akt and cholesterol activation. Glia 2020, 68, 2040–2056. [Google Scholar] [CrossRef] [PubMed]
- Stieber, A.; Gonatas, J.O.; Gonatas, N.K. Aggregates of mutant protein appear progressively in dendrites, in periaxonal processes of oligodendrocytes, and in neuronal and astrocytic perikarya of mice expressing the SOD1(G93A) mutation of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 2000, 177, 114–123. [Google Scholar] [CrossRef]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brannstrom, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Fukuda, Y.; Yoshimura, J.; Toyoda, A.; Kurppa, K.; Moritoyo, H.; Belzil, V.V.; Dion, P.A.; Higasa, K.; Doi, K.; et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am. J. Hum. Genet. 2013, 93, 900–905. [Google Scholar] [CrossRef]
- Sussman, C.R.; Vartanian, T.; Miller, R.H. The ErbB4 neuregulin receptor mediates suppression of oligodendrocyte maturation. J. Neurosci. 2005, 25, 5757–5762. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, B.G.; Agarwal, A.; Sereda, M.W.; Garratt, A.N.; Muller, T.; Wende, H.; Stassart, R.M.; Nawaz, S.; Humml, C.; Velanac, V.; et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 2008, 59, 581–595. [Google Scholar] [CrossRef]
- Takahashi, Y.; Uchino, A.; Shioya, A.; Sano, T.; Matsumoto, C.; Numata-Uematsu, Y.; Nagano, S.; Araki, T.; Murayama, S.; Saito, Y. Altered immunoreactivity of ErbB4, a causative gene product for ALS19, in the spinal cord of patients with sporadic ALS. Neuropathology 2019, 39, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef]
- Magnus, T.; Carmen, J.; Deleon, J.; Xue, H.; Pardo, A.C.; Lepore, A.C.; Mattson, M.P.; Rao, M.S.; Maragakis, N.J. Adult glial precursor proliferation in mutant SOD1G93A mice. Glia 2008, 56, 200–208. [Google Scholar] [CrossRef]
- Eykens, C.; Nonneman, A.; Jensen, C.; Iavarone, A.; Van Damme, P.; Van Den Bosch, L.; Robberecht, W. Conditional deletion of Id2 or Notch1 in oligodendrocyte progenitor cells does not ameliorate disease outcome in SOD1(G93A) mice. Neurobiol. Aging 2018, 68, 1–4. [Google Scholar] [CrossRef]
- Liu, C.; Li, D.; Lv, C.; Gao, Z.; Qi, Y.; Wu, H.; Tian, Y.; Guo, Y. Activation of the Notch Signaling Pathway and Cellular Localization of Notch Signaling Molecules in the Spinal Cord of SOD1-G93A ALS Model Mice. Neuroscience 2020, 432, 84–93. [Google Scholar] [CrossRef]
- Gonzalez, D.; Brandan, E. CTGF/CCN2 from Skeletal Muscle to Nervous System: Impact on Neurodegenerative Diseases. Mol. Neurobiol. 2019, 56, 5911–5916. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sun, Y.; Ling, S.C.; Ferraiuolo, L.; McAlonis-Downes, M.; Zou, Y.; Drenner, K.; Wang, Y.; Ditsworth, D.; Tokunaga, S.; et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. USA 2015, 112, E6993–E7002. [Google Scholar] [CrossRef] [PubMed]
- Falcao, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jakel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; Ffrench-Constant, C.; et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 3887. [Google Scholar] [CrossRef]
- Livesey, M.R.; Magnani, D.; Cleary, E.M.; Vasistha, N.A.; James, O.T.; Selvaraj, B.T.; Burr, K.; Story, D.; Shaw, C.E.; Kind, P.C.; et al. Maturation and electrophysiological properties of human pluripotent stem cell-derived oligodendrocytes. Stem. Cells 2016, 34, 1040–1053. [Google Scholar] [CrossRef]
- Langseth, A.J.; Kim, J.; Ugolino, J.E.; Shah, Y.; Hwang, H.Y.; Wang, J.; Bergles, D.E.; Brown, S.P. Cell-type specific differences in promoter activity of the ALS-linked C9orf72 mouse ortholog. Sci. Rep. 2017, 7, 5685. [Google Scholar] [CrossRef] [PubMed]
- Andres-Benito, P.; Moreno, J.; Aso, E.; Povedano, M.; Ferrer, I. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: Implications in frontotemporal lobar degeneration. Aging 2017, 9, 823–851. [Google Scholar] [CrossRef]
- Wu, Y.J.; Tang, Y.F.; Xiao, Z.C.; Bao, Z.M.; He, B.P. NG2 cells response to axonal alteration in the spinal cord white matter in mice with genetic disruption of neurofilament light subunit expression. Mol. Neurodegener. 2008, 3, 18. [Google Scholar] [CrossRef]
- Bogaert, E.; d’Ydewalle, C.; Van Den Bosch, L. Amyotrophic lateral sclerosis and excitotoxicity: From pathological mechanism to therapeutic target. CNS Neurol. Disord. Drug Targets 2010, 9, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Montibeller, L.; Tan, L.Y.; Kim, J.K.; Paul, P.; de Belleroche, J. Tissue-selective regulation of protein homeostasis and unfolded protein response signaling in sporadic ALS. J. Cell Mol. Med. 2020, 24, 6055–6069. [Google Scholar] [CrossRef]
- Bond, L.; Bernhardt, K.; Madria, P.; Sorrentino, K.; Scelsi, H.; Mitchell, C.S. A Metadata Analysis of Oxidative Stress Etiology in Preclinical Amyotrophic Lateral Sclerosis: Benefits of Antioxidant Therapy. Front. Neurosci. 2018, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef] [PubMed]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, J.E.; Shkreta, L.; Moszczynski, A.J.; Sidibe, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J.; et al. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Robin Highley, J.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Mandler, M.D.; Ku, L.; Feng, Y. A cytoplasmic quaking I isoform regulates the hnRNP F/H-dependent alternative splicing pathway in myelinating glia. Nucleic Acids Res. 2014, 42, 7319–7329. [Google Scholar] [CrossRef]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and microglia as major players of myelin production in normal and pathological conditions. Front. Cell. Neurosci. 2020, 14, 79. [Google Scholar] [CrossRef]
- Clemente, D.; Ortega, M.C.; Melero-Jerez, C.; de Castro, F. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front. Cell. Neurosci. 2013, 7, 268. [Google Scholar] [CrossRef]
- Li, J.; Zhang, L.; Chu, Y.; Namaka, M.; Deng, B.; Kong, J.; Bi, X. Astrocytes in Oligodendrocyte Lineage Development and White Matter Pathology. Front. Cell. Neurosci. 2016, 10, 119. [Google Scholar] [CrossRef]
- Almad, A.A.; Doreswamy, A.; Gross, S.K.; Richard, J.P.; Huo, Y.; Haughey, N.; Maragakis, N.J. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 2016, 64, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Masaki, K.; Yamasaki, R.; Imamura, S.; Suzuki, S.O.; Hayashi, S.; Sato, S.; Nagara, Y.; Kawamura, M.F.; Kira, J. Extensive dysregulations of oligodendrocytic and astrocytic connexins are associated with disease progression in an amyotrophic lateral sclerosis mouse model. J. Neuroinflammation 2014, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins are critical for normal myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Davies, C.L.; Miron, V.E. Microglia: Origins, homeostasis, and roles in myelin repair. Curr. Opin. Neurobiol. 2017, 47, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Lorente Pons, A.; Higginbottom, A.; Cooper-Knock, J.; Alrafiah, A.; Alofi, E.; Kirby, J.; Shaw, P.J.; Wood, J.D.; Highley, J.R. Oligodendrocyte pathology exceeds axonal pathology in white matter in human amyotrophic lateral sclerosis. J. Pathol. 2020, 251, 262–271. [Google Scholar] [CrossRef]
- Kim, S.; Chung, A.Y.; Na, J.E.; Lee, S.J.; Jeong, S.H.; Kim, E.; Sun, W.; Rhyu, I.J.; Park, H.C. Myelin degeneration induced by mutant superoxide dismutase 1 accumulation promotes amyotrophic lateral sclerosis. Glia 2019, 67, 1910–1921. [Google Scholar] [CrossRef]
- Jaarsma, D.; Teuling, E.; Haasdijk, E.D.; De Zeeuw, C.I.; Hoogenraad, C.C. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 2008, 28, 2075–2088. [Google Scholar] [CrossRef]
- Fatima, M.; Tan, R.; Halliday, G.M.; Kril, J.J. Spread of pathology in amyotrophic lateral sclerosis: Assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol. Commun. 2015, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.V.; Fenton, W.A.; McGrath, J.; Horwich, A.L. Transfer of pathogenic and nonpathogenic cytosolic proteins between spinal cord motor neurons in vivo in chimeric mice. Proc. Natl. Acad. Sci. USA 2017, 114, E3139–E3148. [Google Scholar] [CrossRef]
- Oluich, L.J.; Stratton, J.A.; Xing, Y.L.; Ng, S.W.; Cate, H.S.; Sah, P.; Windels, F.; Kilpatrick, T.J.; Merson, T.D. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J. Neurosci. 2012, 32, 8317–8330. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Cooksey, E.; Duncan, I.D. Myelin loss does not lead to axonal degeneration in a long-lived model of chronic demyelination. J. Neurosci. 2013, 33, 2718–2727. [Google Scholar] [CrossRef]
- Barros, L.F. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013, 36, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Asahina, N.; Kitano, S.; Kino, Y. A Comprehensive Profile of ChIP-Seq-Based Olig2 Target Genes in Motor Neuron Progenitor Cells Suggests the Possible Involvement of Olig2 in the Pathogenesis of Amyotrophic Lateral Sclerosis. J. Cent. Nerv. Syst. Dis. 2015, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Starikov, L.; Kottmann, A.H. Diminished Ventral Oligodendrocyte Precursor Generation Results in the Subsequent Over-production of Dorsal Oligodendrocyte Precursors of Aberrant Morphology and Function. Neuroscience 2020, 450, 15–28. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Filipi, T.; Hermanova, Z.; Tureckova, J.; Vanatko, O.; Anderova, A.M. Glial Cells-The Strategic Targets in Amyotrophic Lateral Sclerosis Treatment. J. Clin. Med. 2020, 9, 261. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Nunez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernandez, M.; Marin, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS-A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef]
- Karagiannis, P.; Inoue, H. ALS, a cellular whodunit on motor neuron degeneration. Mol. Cell. Neurosci. 2020, 107, 103524. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Jiang, J.; Cleveland, D.W. Bidirectional Transcriptional Inhibition as Therapy for ALS/FTD Caused by Repeat Expansion in C9orf72. Neuron 2016, 92, 1160–1163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Y.; Dodart, J.C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Chen, P.C.; Hsieh, Y.C.; Huang, C.C.; Hu, C.J. Tamoxifen for amyotrophic lateral sclerosis: A randomized double-blind clinical trial. Medicine 2020, 99, e20423. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonte, C. Actions of the antihistaminergic clemastine on presymptomatic SOD1-G93A mice ameliorate ALS disease progression. J. Neuroinflamm. 2016, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, E.; Bonifacino, T.; Raffaele, S.; Milanese, M.; Morgante, E.; Bonanno, G.; Abbracchio, M.P.; Fumagalli, M. Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice. Int. J. Mol. Sci. 2020, 21, 2395. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Ruiz-Soto, M.; Berciano, M.T.; Berciano, J.; Lafarga, M. Neuroprotective Effect of Bexarotene in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2015, 9, 250. [Google Scholar] [CrossRef]
- Juntas-Morales, R.; Pageot, N.; Bendarraz, A.; Alphandery, S.; Sedel, F.; Seigle, S.; Camu, W. High-dose pharmaceutical grade biotin (MD1003) in amyotrophic lateral sclerosis: A pilot study. EClinicalMedicine 2020, 19, 100254. [Google Scholar] [CrossRef]
- Paganoni, S.; Alshikho, M.J.; Luppino, S.; Chan, J.; Pothier, L.; Schoenfeld, D.; Andres, P.L.; Babu, S.; Zurcher, N.R.; Loggia, M.L.; et al. A pilot trial of RNS60 in amyotrophic lateral sclerosis. Muscle Nerve 2019, 59, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Vallarola, A.; Sironi, F.; Tortarolo, M.; Gatto, N.; De Gioia, R.; Pasetto, L.; De Paola, M.; Mariani, A.; Ghosh, S.; Watson, R.; et al. RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J. Neuroinflamm. 2018, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Espejo-Porras, F.; Garcia-Toscano, L.; Rodriguez-Cueto, C.; Santos-Garcia, I.; de Lago, E.; Fernandez-Ruiz, J. Targeting glial cannabinoid CB2 receptors to delay the progression of the pathological phenotype in TDP-43 (A315T) transgenic mice, a model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2019, 176, 1585–1600. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Mesenchymal Stem Cells: A Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis? Stem Cells Int. 2019, 2019, 3675627. [Google Scholar] [CrossRef]
- Myszczynska, M.; Ferraiuolo, L. New In Vitro Models to Study Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Kunis, G.; Baruch, K.; Miller, O.; Schwartz, M. Immunization with a Myelin-Derived Antigen Activates the Brain’s Choroid Plexus for Recruitment of Immunoregulatory Cells to the CNS and Attenuates Disease Progression in a Mouse Model of ALS. J. Neurosci. 2015, 35, 6381–6393. [Google Scholar] [CrossRef]
- Lubetzki, C.; Zalc, B.; Williams, A.; Stadelmann, C.; Stankoff, B. Remyelination in multiple sclerosis: From basic science to clinical translation. Lancet Neurol. 2020, 19, 678–688. [Google Scholar] [CrossRef]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Bechler, M.E.; Byrne, L.; Ffrench-Constant, C. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. 2015, 25, 2411–2416. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Laouarem, Y.; Kassoussi, A.; Zahaf, A.; Hutteau-Hamel, T.; Mellouk, A.; Bobe, P.; Mattern, C.; Schumacher, M.; Traiffort, E. Functional cooperation of the hedgehog and androgen signaling pathways during developmental and repairing myelination. Glia 2021. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Fancy, S.P.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef]
- Garcia-Leon, J.A.; Kumar, M.; Boon, R.; Chau, D.; One, J.; Wolfs, E.; Eggermont, K.; Berckmans, P.; Gunhanlar, N.; de Vrij, F.; et al. SOX10 Single Transcription Factor-Based Fast and Efficient Generation of Oligodendrocytes from Human Pluripotent Stem Cells. Stem. Cell Rep. 2018, 10, 655–672. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traiffort, E.; Morisset-Lopez, S.; Moussaed, M.; Zahaf, A. Defective Oligodendroglial Lineage and Demyelination in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 3426. https://doi.org/10.3390/ijms22073426
Traiffort E, Morisset-Lopez S, Moussaed M, Zahaf A. Defective Oligodendroglial Lineage and Demyelination in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(7):3426. https://doi.org/10.3390/ijms22073426
Chicago/Turabian StyleTraiffort, Elisabeth, Séverine Morisset-Lopez, Mireille Moussaed, and Amina Zahaf. 2021. "Defective Oligodendroglial Lineage and Demyelination in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 7: 3426. https://doi.org/10.3390/ijms22073426
APA StyleTraiffort, E., Morisset-Lopez, S., Moussaed, M., & Zahaf, A. (2021). Defective Oligodendroglial Lineage and Demyelination in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 22(7), 3426. https://doi.org/10.3390/ijms22073426