Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Akt Isoforms: Differences and Uniqueness
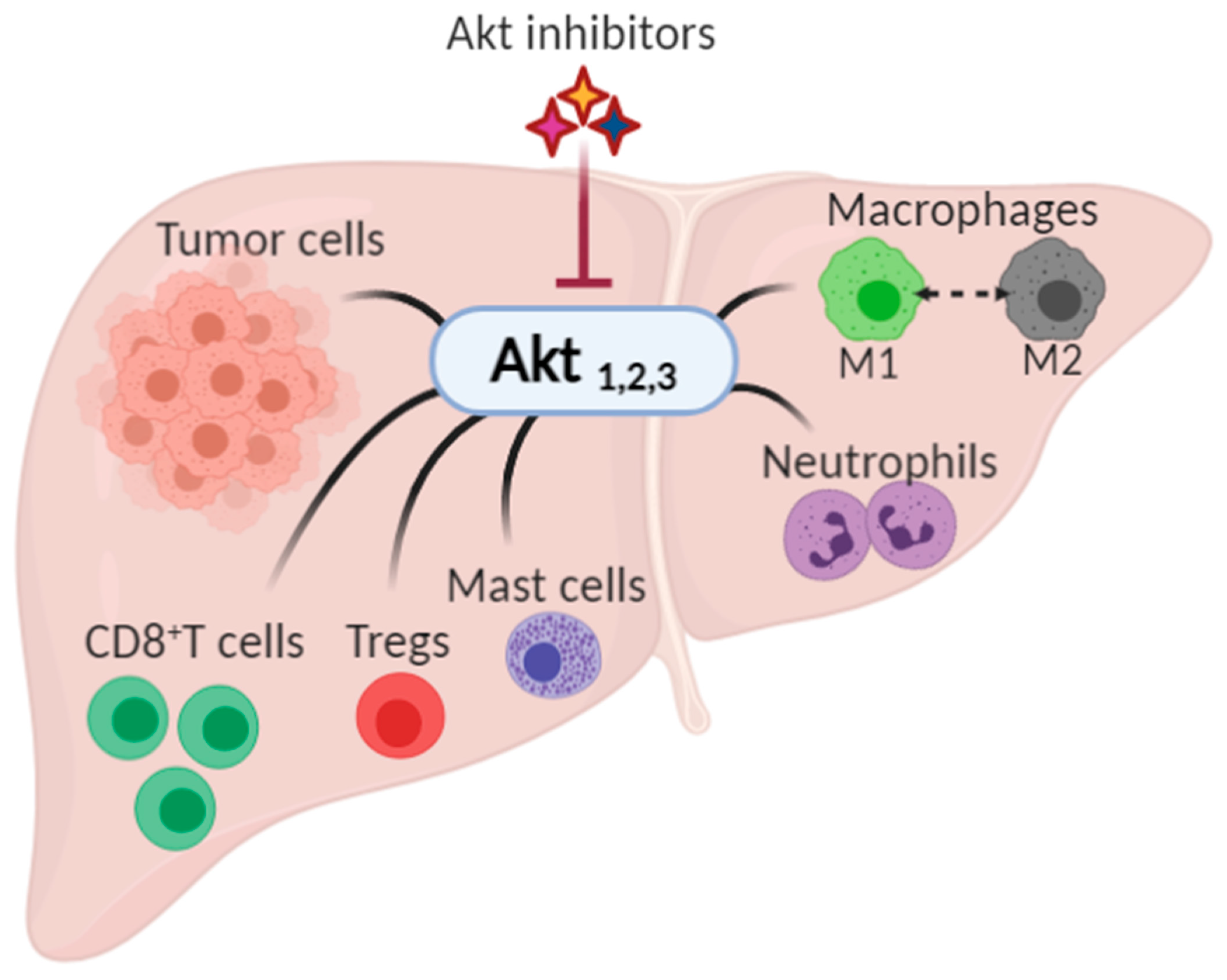
3. Akt in the Development and Progression of Hepatocellular Carcinoma
4. Akt Modulates the Immune Cells
4.1. Akt Regulates the Functions and Fate of T Cells
4.2. Macrophage Polarization
4.3. Other Cells in TME
5. Targeting Akt in the Management of HCC

{kind=link}
| Inhibitor | Mechanism of Action, Structure | Experiment Setup | Antitumor Effect | Effect on TME | Clinical Trial |
|---|---|---|---|---|---|
| GDC-0068 [64,65,66] | ATP competitive AKT inhibitor | Combination of GDC-0068 with Sorafenib in HepG2 and Huh7 sorafenib-resistant cell-lines |
| Not investigated | Phase-I/II including multiple solid tumors treated by GDC-0068 in monotherapy or in association with abiraterone + prednisolone: safe and tolerable in monotherapy or in combination |
| AZD5363 [67,68] | ATP competitive AKT inhibitor | Single agent in HepG2 and Huh-7 HCC cells Combination with FH535 (β-catenin inhibitor) in THH, Hep3B and HepG2 |
| Not investigated | Phase-I, AZD5363 in monotherapy, in multiple advanced solid tumors including liver cancer (NCT01895946): safe and tolerable, no data on antitumor response Phase-I including multiple solid tumors harboring AKT mutations (NCT02465060) |
| ARQ 092 [69,70] | allosteric pan-AKT inhibitor | Single agent and in combination with Sorafenib in a DEN-induced cirrhotic rat model of HCC and in Hep3B, HepG2, Huh-7, PLC/PRF, and HR4 HCC cell lines |
|
| No clinical trial |
| ARQ 751 [71] | allosteric pan-AKT inhibitor Chemical structure of ARQ 751 is currently unavailable. | Single agent and in combination with sorafinib in DEN-induced cirrhotic rat model of HCC |
|
| Phase-Ib (NCT02761694) in solid tumors with PIK3CA/AKT/PTEN mutations including HCC: ongoing |
| MK-2206 [72,73] | allosteric pan-AKT inhibitor | Single agent in Human Huh7, Hep3B, and HepG2 HCC cell lines |
| Not investigated | Phase-II trial, MK-2206 in monotherapy in advanced HCC previously treated (NCT01239355): discontinued due to discouraging results |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.M. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar]
- Ogunwobi, O.O.; Harricharran, T.; Huaman, J.; Galuza, A.; Odumuwagun, O.; Tan, Y.; Ma, G.X.; Nguyen, M.T. Mechanisms of hepatocellular carcinoma progression. World J. Gastroenterol. 2019, 25, 2279–2293. [Google Scholar] [CrossRef] [PubMed]
- Mroweh, M.; Decaens, T.; Marche, P.N.; Macek Jilkova, Z.; Clément, F. Modulating the Crosstalk between the Tumor and Its Microenvironment Using RNA Interference: A Treatment Strategy for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 5250. [Google Scholar] [CrossRef]
- Nault, J.C.; Zucman-Rossi, J. Genetics of hepatocellular carcinoma: The next generation. J. Hepatol. 2014, 60, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Viatour, P. Hepatocellular carcinoma: Old friends and new tricks. Exp. Mol. Med. 2020, 52, 1898–1907. [Google Scholar] [CrossRef]
- Macek-Jilkova, Z.; Malov, S.I.; Kurma, K.; Charrat, C.; Decaens, T.; Peretolchina, N.P.; Marche, P.N.; Malov, I.V.; Yushchuk, N.D. Clinical and Experimental Evaluation of Diagnostic Significance of Alpha-Fetoprotein and Osteopontin at the Early Stage of Hepatocellular Cancer. Bull. Exp. Biol. Med. 2021, 170, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Fan, G.; Wei, X.; Xu, X. Is the era of sorafenib over? A review of the literature. Ther. Adv. Med. Oncol. 2020, 12, 1758835920927602. [Google Scholar] [CrossRef]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Aspord, C.; Decaens, T. Predictive Factors for Response to PD-1/PD-L1 Checkpoint Inhibition in the Field of Hepatocellular Carcinoma: Current Status and Challenges. Cancers 2019, 11, 1554. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Testa, J.R.; Staal, S.P.; Tsichlis, P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, X.; Hay, N. Akt as a target for cancer therapy: More is not always better (lessons from studies in mice). Br. J. Cancer 2017, 117, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ferrín, G.; Guerrero, M.; Amado, V.; Rodríguez-Perálvarez, M.; De la Mata, M. Activation of mTOR Signaling Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef]
- Son, M.K.; Ryu, Y.-L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.-K.; Suh, J.K.; Hong, S.; et al. HS-173, a Novel PI3K Inhibitor, Attenuates the Activation of Hepatic Stellate Cells in Liver Fibrosis. Sci. Rep. 2013, 3, 3470. [Google Scholar] [CrossRef]
- Cai, C.X.; Buddha, H.; Castelino-Prabhu, S.; Zhang, Z.; Britton, R.S.; Bacon, B.R.; Neuschwander-Tetri, B.A. Activation of Insulin-PI3K/Akt-p70S6K Pathway in Hepatic Stellate Cells Contributes to Fibrosis in Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2017, 62, 968–978. [Google Scholar] [CrossRef]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol. 2008, 48, 83–90. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, X.; Yang, M.; Yu, L.; Li, Z.; Lin, N. Identification of AKT kinases as unfavorable prognostic factors for hepatocellular carcinoma by a combination of expression profile, interaction network analysis and clinical validation. Mol. Biosyst. 2014, 10, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xu, M.; Liu, P.; Zhang, S.; Shang, R.; Qiao, Y.; Che, L.; Ribback, S.; Cigliano, A.; Evert, K.; et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 2019, 70, 1600–1613. [Google Scholar] [CrossRef]
- Zhao, J.X.; Yuan, Y.W.; Cai, C.F.; Shen, D.Y.; Chen, M.L.; Ye, F.; Mi, Y.J.; Luo, Q.C.; Cai, W.Y.; Zhang, W.; et al. Aldose reductase interacts with AKT1 to augment hepatic AKT/mTOR signaling and promote hepatocarcinogenesis. Oncotarget 2017, 8, 66987–67000. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Yu, W.; Song, J.; Li, Q.; Wang, C.; Xie, S. Extracellular polyamines-induced proliferation and migration of cancer cells by ODC, SSAT, and Akt1-mediated pathway. Anti-Cancer Drugs 2017, 28, 457–464. [Google Scholar] [CrossRef]
- Delogu, S.; Wang, C.; Cigliano, A.; Utpatel, K.; Sini, M.; Longerich, T.; Waldburger, N.; Breuhahn, K.; Jiang, L.; Ribback, S.; et al. SKP2 cooperates with N-Ras or AKT to induce liver tumor development in mice. Oncotarget 2015, 6, 2222–2234. [Google Scholar] [CrossRef]
- Zhu, L.; Hu, C.; Li, J.; Xue, P.; He, X.; Ge, C.; Qin, W.; Yao, G.; Gu, J. Real-time imaging nuclear translocation of Akt1 in HCC cells. Biochem. Biophys. Res. Commun. 2007, 356, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Meng, X.W.; Sun, X.; Liu, B.R.; Han, M.Z.; Du, Y.J.; Song, Y.Y.; Xu, W. Wnt/β-catenin signaling regulates MAPK and Akt1 expression and growth of hepatocellular carcinoma cells. Neoplasma 2011, 58, 239–244. [Google Scholar] [CrossRef]
- Xu, X.; Sakon, M.; Nagano, H.; Hiraoka, N.; Yamamoto, H.; Hayashi, N.; Dono, K.; Nakamori, S.; Umeshita, K.; Ito, Y.; et al. Akt2 expression correlates with prognosis of human hepatocellular carcinoma. Oncol. Rep. 2004, 11, 25–32. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Zhang, C. STAT3 promotes the proliferation and migration of hepatocellular carcinoma cells by regulating AKT2. Oncol. Lett. 2018, 15, 3333–3338. [Google Scholar] [CrossRef]
- Wang, C.; Che, L.; Hu, J.; Zhang, S.; Jiang, L.; Latte, G.; Demartis, M.I.; Tao, J.; Gui, B.; Pilo, M.G.; et al. Activated mutant forms of PIK3CA cooperate with RasV12 or c-Met to induce liver tumour formation in mice via AKT2/mTORC1 cascade. Liver Int. Off. J. Int. Assoc. Study Liver 2016, 36, 1176–1186. [Google Scholar] [CrossRef]
- Chew, T.W.; Liu, X.J.; Liu, L.; Spitsbergen, J.M.; Gong, Z.; Low, B.C. Crosstalk of Ras and Rho: Activation of RhoA abates Kras-induced liver tumorigenesis in transgenic zebrafish models. Oncogene 2014, 33, 2717–2727. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, W.N.; Chen, X.; Peng, X.D.; Jeon, S.M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, W.; Ran, Y.; Xiong, Y.; Zhong, Z.; Fan, X.; Wang, Z.; Ye, Q. miR-582-5p inhibits proliferation of hepatocellular carcinoma by targeting CDK1 and AKT3. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 8309–8316. [Google Scholar] [CrossRef]
- Ma, Y.; She, X.G.; Ming, Y.Z.; Wan, Q.Q.; Ye, Q.F. MicroRNA‑144 suppresses tumorigenesis of hepatocellular carcinoma by targeting AKT3. Mol. Med. Rep. 2015, 11, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Graupera, M.; Vanhaesebroeck, B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016, 6, 1090–1105. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Elgaaied, A.B.; Chouaib, S. Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk with Tumor Microenvironment. Front. Immunol. 2017, 8, 270. [Google Scholar] [CrossRef]
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Sun, I.H.; Patel, C.H.; Lo, Y.C.; Oh, M.H.; Waickman, A.T.; Tam, A.J.; Blosser, R.L.; Wen, J.; Delgoffe, G.M.; et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat. Immunol. 2016, 17, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Lerner, C.G.; Schwartz, R.H. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J. Immunol. 1999, 162, 2775–2784. [Google Scholar] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Finlay, D.; Feijoo, C.; Cornish, G.H.; Gray, A.; Ager, A.; Okkenhaug, K.; Hagenbeek, T.J.; Spits, H.; Cantrell, D.A. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 2008, 9, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Zeng, Q.; Tang, N.; Guo, H.; Tang, R.; Yin, W.; Wang, A.; Tang, H.; Zhou, J.; Xie, H.; et al. Akt-1 and Akt-2 Differentially Regulate the Development of Experimental Autoimmune Encephalomyelitis by Controlling Proliferation of Thymus-Derived Regulatory T Cells. J. Immunol. 2019, 202, 1441–1452. [Google Scholar] [CrossRef]
- DuBois, J.C.; Ray, A.K.; Gruber, R.C.; Zhang, Y.; Aflakpui, R.; Macian-Juan, F.; Shafit-Zagardo, B. Akt3-Mediated Protection Against Inflammatory Demyelinating Disease. Front. Immunol. 2019, 10, 1738. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Weichhart, T.; Saemann, M.D. The PI3K/Akt/mTOR pathway in innate immune cells: Emerging therapeutic applications. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), iii70-4. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Schifferle, R.E.; Cuesta, N.; Vogel, S.N.; Katz, J.; Michalek, S.M. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J. Immunol. 2003, 171, 717–725. [Google Scholar] [CrossRef]
- Rocher, C.; Singla, D.K. SMAD-PI3K-Akt-mTOR pathway mediates BMP-7 polarization of monocytes into M2 macrophages. PLoS ONE 2013, 8, e84009. [Google Scholar] [CrossRef]
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Sly, L.M.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP represses the generation of alternatively activated macrophages. Immunity 2005, 23, 361–374. [Google Scholar] [CrossRef]
- Araki, K.; Ellebedy, A.H.; Ahmed, R. TOR in the immune system. Curr. Opin Cell Biol. 2011, 23, 707–715. [Google Scholar] [CrossRef]
- Mercalli, A.; Calavita, I.; Dugnani, E.; Citro, A.; Cantarelli, E.; Nano, R.; Melzi, R.; Maffi, P.; Secchi, A.; Sordi, V.; et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology 2013, 140, 179–190. [Google Scholar] [CrossRef]
- Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Huber, M.; Kalis, C.; Keck, S.; Galanos, C.; et al. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005, 6, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Strassheim, D.; Asehnoune, K.; Park, J.S.; Kim, J.Y.; He, Q.; Richter, D.; Kuhn, K.; Mitra, S.; Abraham, E. Phosphoinositide 3-kinase and Akt occupy central roles in inflammatory responses of Toll-like receptor 2-stimulated neutrophils. J. Immunol. 2004, 172, 5727–5733. [Google Scholar] [CrossRef]
- Schaeffer, V.; Arbabi, S.; Garcia, I.A.; Knoll, M.L.; Cuschieri, J.; Bulger, E.M.; Maier, R.V. Role of the mTOR pathway in LPS-activated monocytes: Influence of hypertonic saline. J. Surg Res. 2011, 171, 769–776. [Google Scholar] [CrossRef]
- Arranz, A.; Doxaki, C.; Vergadi, E.; de la Torre, Y.M.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef]
- Chen, J.; Tang, H.; Hay, N.; Xu, J.; Ye, R.D. Akt isoforms differentially regulate neutrophil functions. Blood 2010, 115, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, C.; Xie, H.; Fan, Y.; Yang, Z.; Ma, J.; He, D.; Li, L. Infiltrating mast cells promote renal cell carcinoma angiogenesis by modulating PI3K→ AKT→ GSK3β→ AM signaling. Oncogene 2017, 36, 2879–2888. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Garrett, M.D.; Walton, M.I.; Raynaud, F.; de Bono, J.S.; Workman, P. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharmacol. 2008, 8, 393–412. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Barnett, S.F.; Yaroschak, M.; Bilodeau, M.T.; Layton, M.E. Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr. Top. Med. Chem. 2007, 7, 1349–1363. [Google Scholar] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Landel, I.; Quambusch, L.; Depta, L.; Rauh, D. Spotlight on AKT: Current Therapeutic Challenges. ACS Med. Chem. Lett. 2020, 11, 225–227. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef]
- Doi, T.; Fujiwara, Y.; Matsubara, N.; Tomomatsu, J.; Iwasa, S.; Tanaka, A.; Endo-Tsukude, C.; Nakagawa, S.; Takahashi, S. Phase I study of ipatasertib as a single agent and in combination with abiraterone plus prednisolone in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2019, 84, 393–404. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Faheem, A.; Sun, T.; Li, C.; Li, Z.; Zhao, D.; Wu, C.; Liu, J. A novel AKT inhibitor, AZD5363, inhibits phosphorylation of AKT downstream molecules, and activates phosphorylation of mTOR and SMG-1 dependent on the liver cancer cell type. Oncol. Lett. 2016, 11, 1685–1692. [Google Scholar] [CrossRef]
- Dean, E.; Banerji, U.; Schellens, J.H.M.; Krebs, M.G.; Jimenez, B.; van Brummelen, E.; Bailey, C.; Casson, E.; Cripps, D.; Cullberg, M.; et al. A Phase 1, open-label, multicentre study to compare the capsule and tablet formulations of AZD5363 and explore the effect of food on the pharmacokinetic exposure, safety and tolerability of AZD5363 in patients with advanced solid malignancies: OAK. Cancer Chemother. Pharmacol. 2018, 81, 873–883. [Google Scholar] [CrossRef]
- Roth, G.S.; Jilkova, Z.M.; Kuyucu, A.Z.; Kurma, K.; Pour, S.T.A.; Abbadessa, G.; Yu, Y.; Busser, B.; Marche, P.N.; Leroy, V.; et al. Efficacy of AKT Inhibitor ARQ 092 Compared with Sorafenib in a Cirrhotic Rat Model with Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Jilkova, Z.M.; Kuyucu, A.Z.; Kurma, K.; Pour, S.T.A.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Schwartz, B.; Sturm, N.; Marche, P.N.; et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget 2018, 9, 11145–11158. [Google Scholar] [CrossRef]
- Kurma, K.; Jilkova, Z.M.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Marche, P.; Decaens, T. Effect of novel AKT inhibitor ARQ 751 as single agent and its combination with sorafenib on hepatocellular carcinoma in a cirrhotic rat model. J. Hepatol. 2017, 66, S459–S460. [Google Scholar]
- Wilson, J.M.; Kunnimalaiyaan, S.; Gamblin, T.C.; Kunnimalaiyaan, M. MK2206 inhibits hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest. J. Surg. Res. 2014, 191, 280–285. [Google Scholar] [CrossRef]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef]
- Patra, T.; Meyer, K.; Ray, R.B.; Ray, R. A combination of AZD5363 and FH5363 induces lethal autophagy in transformed hepatocytes. Cell Death Dis. 2020, 11, 540. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8+ T-cell activity. J. Immunother. Cancer 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective Inhibition of Regulatory T Cells by Targeting the PI3K–Akt Pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Searle, E.J.; Telfer, B.A.; Mukherjee, D.; Forster, D.M.; Davies, B.R.; Williams, K.J.; Stratford, I.J.; Illidge, T.M. Akt inhibition improves long-term tumour control following radiotherapy by altering the microenvironment. Embo Mol. Med. 2017, 9, 1646–1659. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Kurma, K.; Decaens, T. Animal Models of Hepatocellular Carcinoma: The Role of Immune System and Tumor Microenvironment. Cancers 2019, 11, 1487. [Google Scholar] [CrossRef]
- Marks, D.K.; Gartrell, R.D.; El Asmar, M.; Boboila, S.; Hart, T.; Lu, Y.; Pan, Q.; Yu, J.; Hibshoosh, H.; Guo, H.; et al. Akt Inhibition Is Associated With Favorable Immune Profile Changes Within the Tumor Microenvironment of Hormone Receptor Positive, HER2 Negative Breast Cancer. Front. Oncol. 2020, 10, 968. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroweh, M.; Roth, G.; Decaens, T.; Marche, P.N.; Lerat, H.; Macek Jílková, Z. Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 1794. https://doi.org/10.3390/ijms22041794
Mroweh M, Roth G, Decaens T, Marche PN, Lerat H, Macek Jílková Z. Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(4):1794. https://doi.org/10.3390/ijms22041794
Chicago/Turabian StyleMroweh, Mariam, Gaël Roth, Thomas Decaens, Patrice N. Marche, Hervé Lerat, and Zuzana Macek Jílková. 2021. "Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 4: 1794. https://doi.org/10.3390/ijms22041794
APA StyleMroweh, M., Roth, G., Decaens, T., Marche, P. N., Lerat, H., & Macek Jílková, Z. (2021). Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment. International Journal of Molecular Sciences, 22(4), 1794. https://doi.org/10.3390/ijms22041794