
The Role of Non-Catalytic Domains of Hrp3 in Nucleosome Remodeling


Abstract
1. Introduction
2. Results
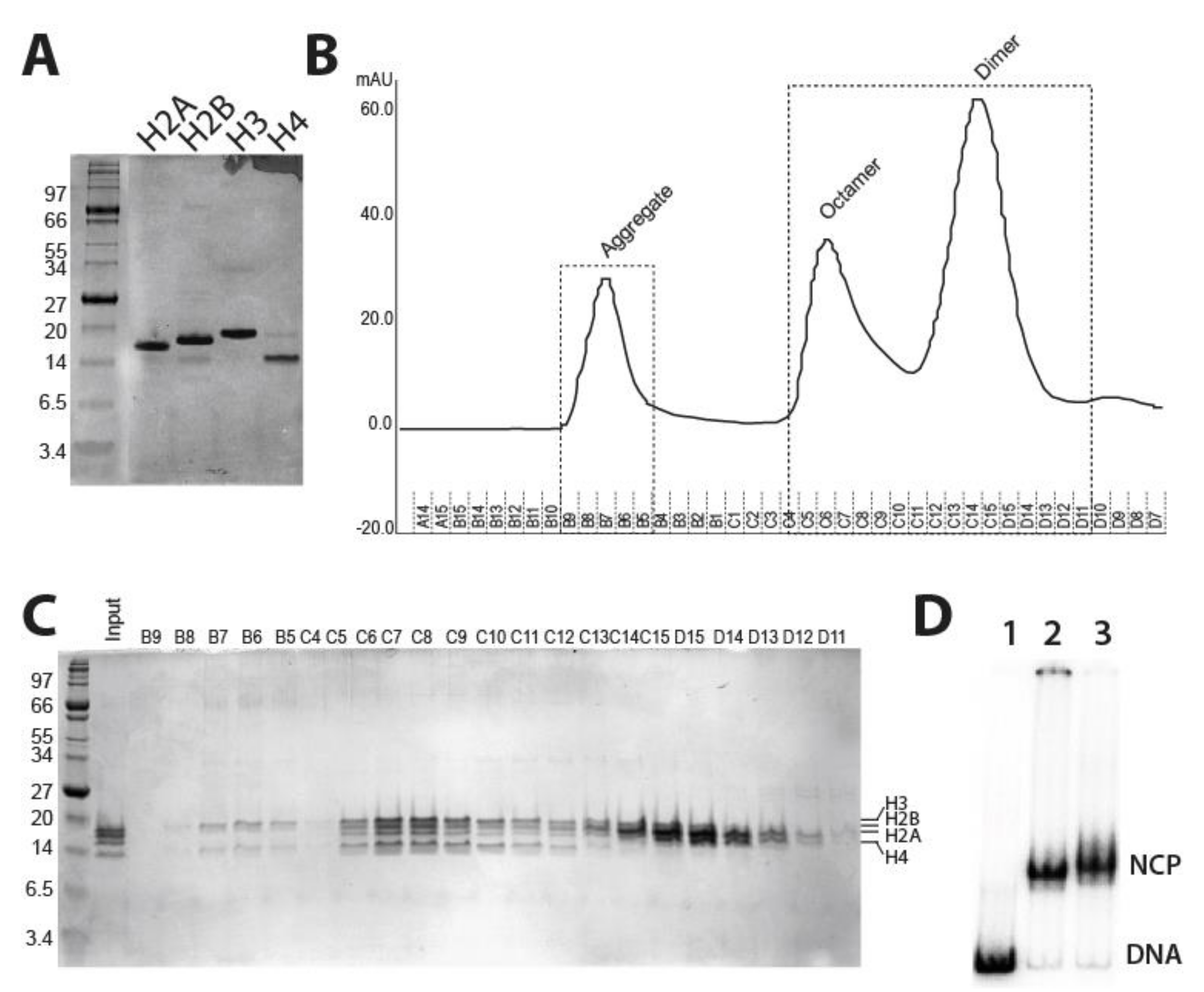
2.1. Purification of Fission Yeast Recombinant Histones and Reconstitution of Mononucleosomes
2.2. Generation of Hrp3 Mutants and Affinity Purification of WT and Mutant Proteins from Fission Yeast
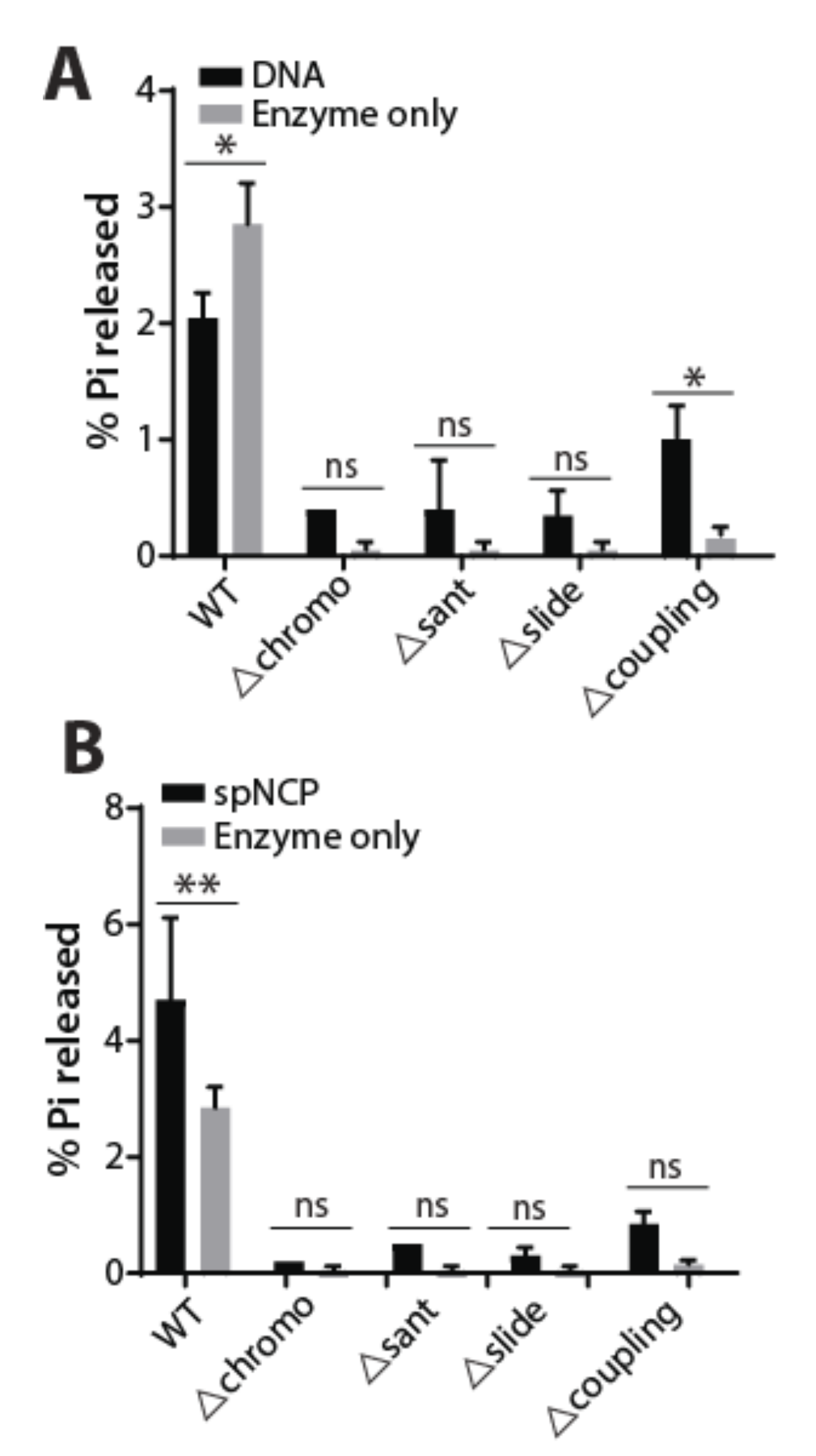
2.3. ATPase Activity of WT and Mutant Hrp3 with DNA and Nucleosomal Substrates
3. Discussion
4. Materials and Methods
4.1. Fission Yeast Strains
4.2. Expression of Histone Proteins
4.3. Preparation of Inclusion Bodies
4.4. Purification of Fission Yeast Histone Proteins
4.5. Histone Octamer Refolding and Purification
4.6. Nucleosome Reconstitution
4.7. Affinity-Purification of Wild Type and Mutant Hrp3 Proteins
4.8. In Vitro ATPase Activity Assay
4.9. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Chromosomal DNA and its packaging in the chromatin fiber. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu Rev. Biochem 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Kulaeva, O.I.; Gaykalova, D.A.; Pestov, N.A.; Golovastov, V.V.; Vassylyev, D.G.; Artsimovitch, I.; Studitsky, V.M. Mechanism of chromatin remodeling and recovery during passage of rna polymerase ii. Nat. Struct. Mol. Biol. 2009, 16, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Vignali, M.; Hassan, A.H.; Neely, K.E.; Workman, J.L. Atp-dependent chromatin-remodeling complexes. Mol. Cell Biol. 2000, 20, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Marfella, C.G.A.; Imbalzano, A.N. The chd family of chromatin remodelers. Mutat. Res. Fund Mol. M 2007, 618, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., 3rd; Millhouse, S.; Chen, C.F.; Lewis, B.A.; Erdjument-Bromage, H.; Tempst, P.; Manley, J.L.; Reinberg, D. Recognition of trimethylated histone h3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mrna splicing. Mol. Cell 2007, 28, 665–676. [Google Scholar] [CrossRef]
- Park, D.; Shivram, H.; Iyer, V.R. Chd1 co-localizes with early transcription elongation factors independently of h3k36 methylation and releases stalled rna polymerase ii at introns. Epigenetics Chromatin 2014, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of chd1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. Embo Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodelling factor chd1 in pten-deficient cancer. Nature 2017, 542, 484–488. [Google Scholar] [CrossRef]
- Schoberleitner, I.; Mutti, A.; Sah, A.; Wille, A.; Gimeno-Valiente, F.; Piatti, P.; Kharitonova, M.; Torres, L.; López-Rodas, G.; Liu, J.J.; et al. Role for chromatin remodeling factor chd1 in learning and memory. Front. Mol. Neurosci. 2019, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Lennartsson, A.; Ekwall, K. The roles of snf2/swi2 nucleosome remodeling enzymes in blood cell differentiation and leukemia. Biomed. Res. Int. 2015, 2015, 347571. [Google Scholar] [CrossRef]
- Siggens, L.; Cordeddu, L.; Ronnerblad, M.; Lennartsson, A.; Ekwall, K. Transcription-coupled recruitment of human chd1 and chd2 influences chromatin accessibility and histone h3 and h3.3 occupancy at active chromatin regions. Epigenetics Chromatin 2015, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Lusser, A.; Urwin, D.L.; Kadonaga, J.T. Distinct activities of chd1 and acf in atp-dependent chromatin assembly. Nat. Struct. Mol. Biol. 2005, 12, 160–166. [Google Scholar] [CrossRef]
- Ocampo, J.; Chereji, R.V.; Eriksson, P.R.; Clark, D.J. The isw1 and chd1 atp-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res. 2016, 44, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Hennig, B.P.; Bendrin, K.; Zhou, Y.; Fischer, T. Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. Embo Rep. 2012, 13, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hauk, G.; McKnight, J.N.; Nodelman, I.M.; Bowman, G.D. The chromodomains of the chd1 chromatin remodeler regulate DNA access to the atpase motor. Mol. Cell 2010, 39, 711–723. [Google Scholar] [CrossRef]
- Torigoe, S.E.; Patel, A.; Khuong, M.T.; Bowman, G.D.; Kadonaga, J.T. Atp-dependent chromatin assembly is functionally distinct from chromatin remodeling. Elife 2013, 2, e00863. [Google Scholar] [CrossRef]
- Ryan, D.P.; Sundaramoorthy, R.; Martin, D.; Singh, V.; Owen-Hughes, T. The DNA-binding domain of the chd1 chromatin-remodelling enzyme contains sant and slide domains. Embo J. 2011, 30, 2596–2609. [Google Scholar] [CrossRef]
- Patel, A.; McKnight, J.N.; Genzor, P.; Bowman, G.D. Identification of residues in chromodomain helicase DNA-binding protein 1 (chd1) required for coupling atp hydrolysis to nucleosome sliding. J. Biol. Chem. 2011, 286, 43984–43993. [Google Scholar] [CrossRef]
- Farnung, L.; Vos, S.M.; Wigge, C.; Cramer, P. Nucleosome-chd1 structure and implications for chromatin remodelling. Nature 2017, 550, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Olsson, I.; Bjerling, P. Advancing our understanding of functional genome organisation through studies in the fission yeast. Curr. Genet. 2011, 57, 1–12. [Google Scholar] [CrossRef]
- Pointner, J.; Persson, J.; Prasad, P.; Norman-Axelsson, U.; Stralfors, A.; Khorosjutina, O.; Krietenstein, N.; Svensson, J.P.; Ekwall, K.; Korber, P. Chd1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in s. Pombe. Embo J. 2012, 31, 4388–4403. [Google Scholar] [CrossRef]
- Adachi, A.; Senmatsu, S.; Asada, R.; Abe, T.; Hoffman, C.S.; Ohta, K.; Hirota, K. Interplay between chromatin modulators and histone acetylation regulates the formation of accessible chromatin in the upstream regulatory region of fission yeast fbp1. Genes Genet. Syst. 2018, 92, 267–276. [Google Scholar] [CrossRef]
- Nodelman, I.M.; Bowman, G.D. Nucleosome sliding by chd1 does not require rigid coupling between DNA-binding and atpase domains. Embo Rep. 2013, 14, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Allshire, R.C.; Ekwall, K. Epigenetic regulation of chromatin states in schizosaccharomyces pombe. Cold Spring Harb. Perspect. Biol. 2015, 7, a018770. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Nagakura, W.; Tanaka, H.; Kujirai, T.; Chikashige, Y.; Haraguchi, T.; Hiraoka, Y.; Kurumizaka, H. In vitro reconstitution and biochemical analyses of the schizosaccharomyces pombe nucleosome. Biochem. Biophys. Res. Commun. 2017, 482, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Rechsteiner, T.J.; Richmond, T.J. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999, 304, 3–19. [Google Scholar]
- McKnight, J.N.; Jenkins, K.R.; Nodelman, I.M.; Escobar, T.; Bowman, G.D. Extranucleosomal DNA binding directs nucleosome sliding by chd1. Mol. Cell. Biol. 2011, 31, 4746–4759. [Google Scholar] [CrossRef]
- Sharma, A.; Jenkins, K.R.; Héroux, A.; Bowman, G.D. Crystal structure of the chromodomain helicase DNA-binding protein 1 (chd1) DNA-binding domain in complex with DNA. J. Biol. Chem. 2011, 286, 42099–42104. [Google Scholar] [CrossRef]
- Dyer, P.N.; Edayathumangalam, R.S.; White, C.L.; Bao, Y.; Chakravarthy, S.; Muthurajan, U.M.; Luger, K. Reconstitution of nucleosome core particles from recombinant histones and DNA. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 375, pp. 23–44. [Google Scholar]
- Rogge, R.A.; Kalashnikova, A.A.; Muthurajan, U.M.; Porter-Goff, M.E.; Luger, K.; Hansen, J.C. Assembly of nucleosomal arrays from recombinant core histones and nucleosome positioning DNA. JoVE 2013, 2013, e50354. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain Name | Genotype | Source |
|---|---|---|
| Hu2402 | h- hrp1D::ura4(1/2 gene deleted), ade6-M210 leu1-32 ura4-D18 Hrp3_TAP | This study |
| Hu3060 | h- hrp1D::ura4(1/2 gene deleted), ade6-M210 leu1-32 ura4-D18 Hrp3 chromodomain∆ _ TAP | This study |
| Hu3061 | h- hrp1D::ura4(1/2 gene deleted), ade6-M210 leu1-32 ura4-D18 Hrp3 ∆sant _ TAP | This study |
| Hu3062 | h- hrp1D::ura4(1/2 gene deleted), ade6-M210 leu1-32 ura4-D18 Hrp3 ∆slide _ TAP | This study |
| Hu3063 | h- hrp1D::ura4(1/2 gene deleted), ade6-M210 leu1-32 ura4-D18 Hrp3 coupling region∆ _ TAP | This study |
| Hu2204 | h- Hrp3::leu2+leu1-32 hrp1_2xFLAGKanMX | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, W.; Prasad, P.; Lennartsson, A.; Ekwall, K. The Role of Non-Catalytic Domains of Hrp3 in Nucleosome Remodeling. Int. J. Mol. Sci. 2021, 22, 1793. https://doi.org/10.3390/ijms22041793
Dong W, Prasad P, Lennartsson A, Ekwall K. The Role of Non-Catalytic Domains of Hrp3 in Nucleosome Remodeling. International Journal of Molecular Sciences. 2021; 22(4):1793. https://doi.org/10.3390/ijms22041793
Chicago/Turabian StyleDong, Wenbo, Punit Prasad, Andreas Lennartsson, and Karl Ekwall. 2021. "The Role of Non-Catalytic Domains of Hrp3 in Nucleosome Remodeling" International Journal of Molecular Sciences 22, no. 4: 1793. https://doi.org/10.3390/ijms22041793
APA StyleDong, W., Prasad, P., Lennartsson, A., & Ekwall, K. (2021). The Role of Non-Catalytic Domains of Hrp3 in Nucleosome Remodeling. International Journal of Molecular Sciences, 22(4), 1793. https://doi.org/10.3390/ijms22041793