
Therapeutic Targeting of Fumaryl Acetoacetate Hydrolase in Hereditary Tyrosinemia Type I

 , , ,
, , ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Dimerization Interface Is Druggable
2.2. Small Molecules to Stabilize the Dimer Conformation of FAH
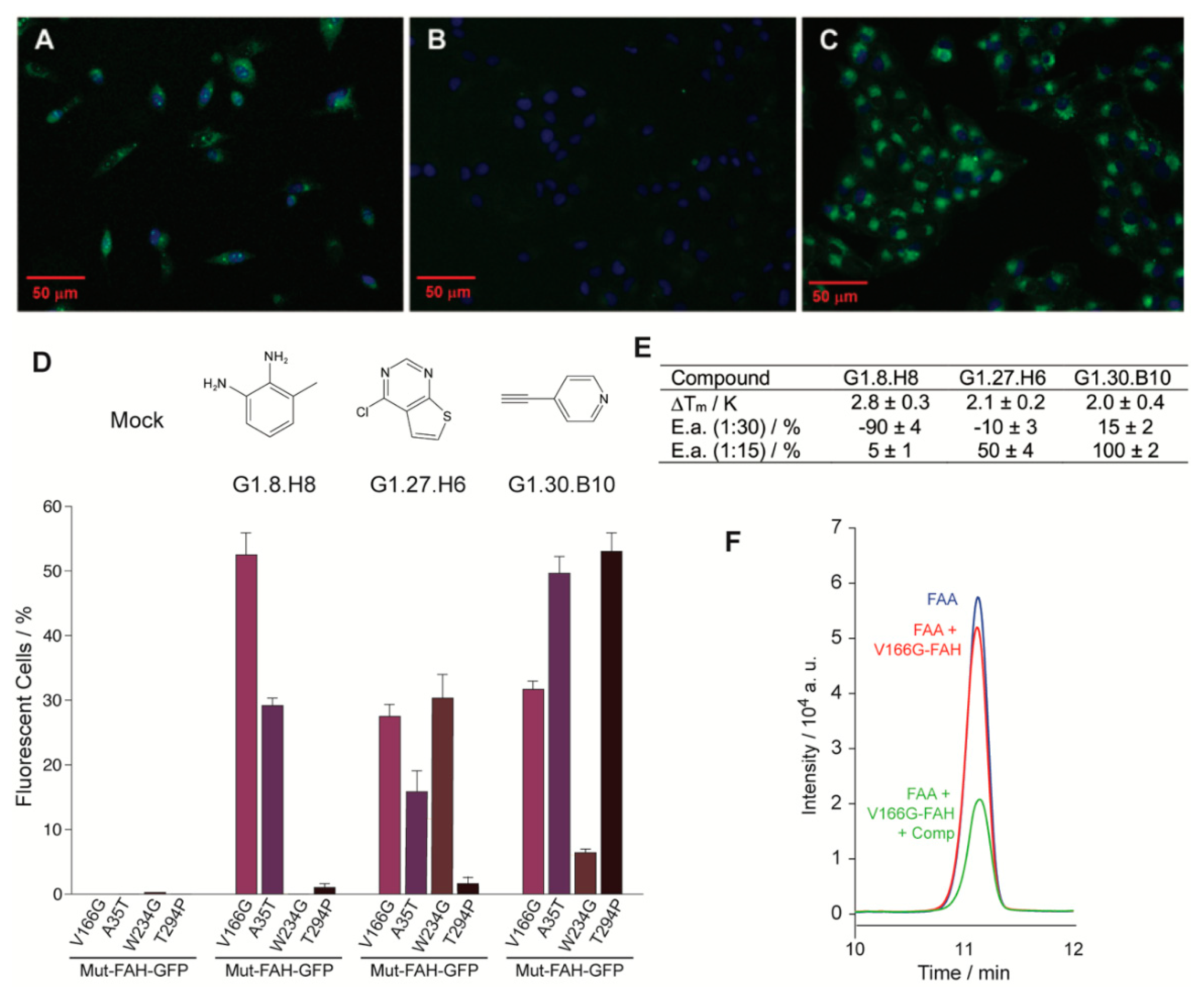
2.3. Functional Screening to Improve Intracellular FAH Homeostasis
2.4. Organic Molecules Modulate FAH Catalytic Activity In Vitro
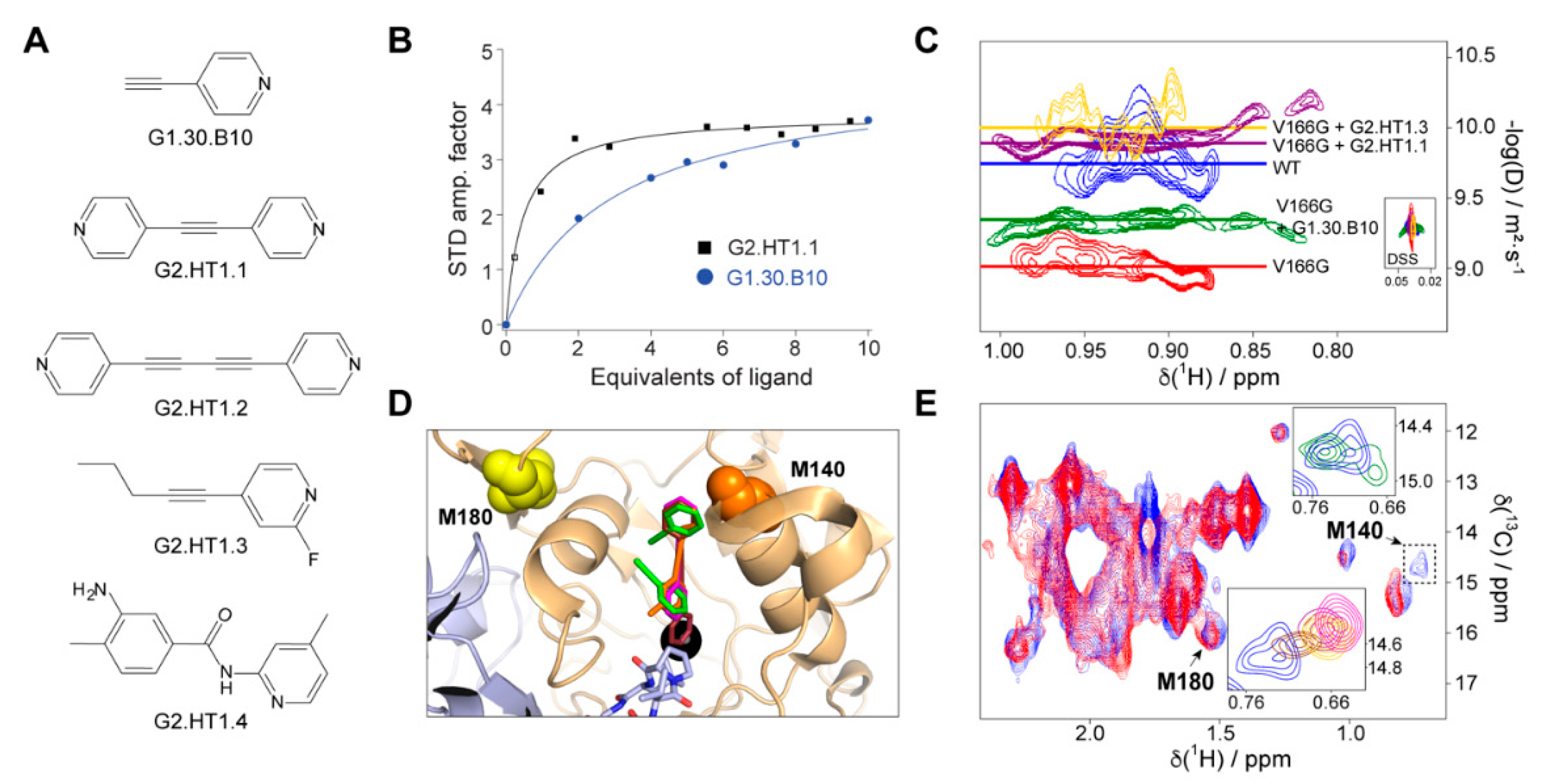
2.5. The Compound G1.30.B10 Stabilizes the Dimeric Species of FAH
2.6. Structural Design of a Second Generation of FAH Activators
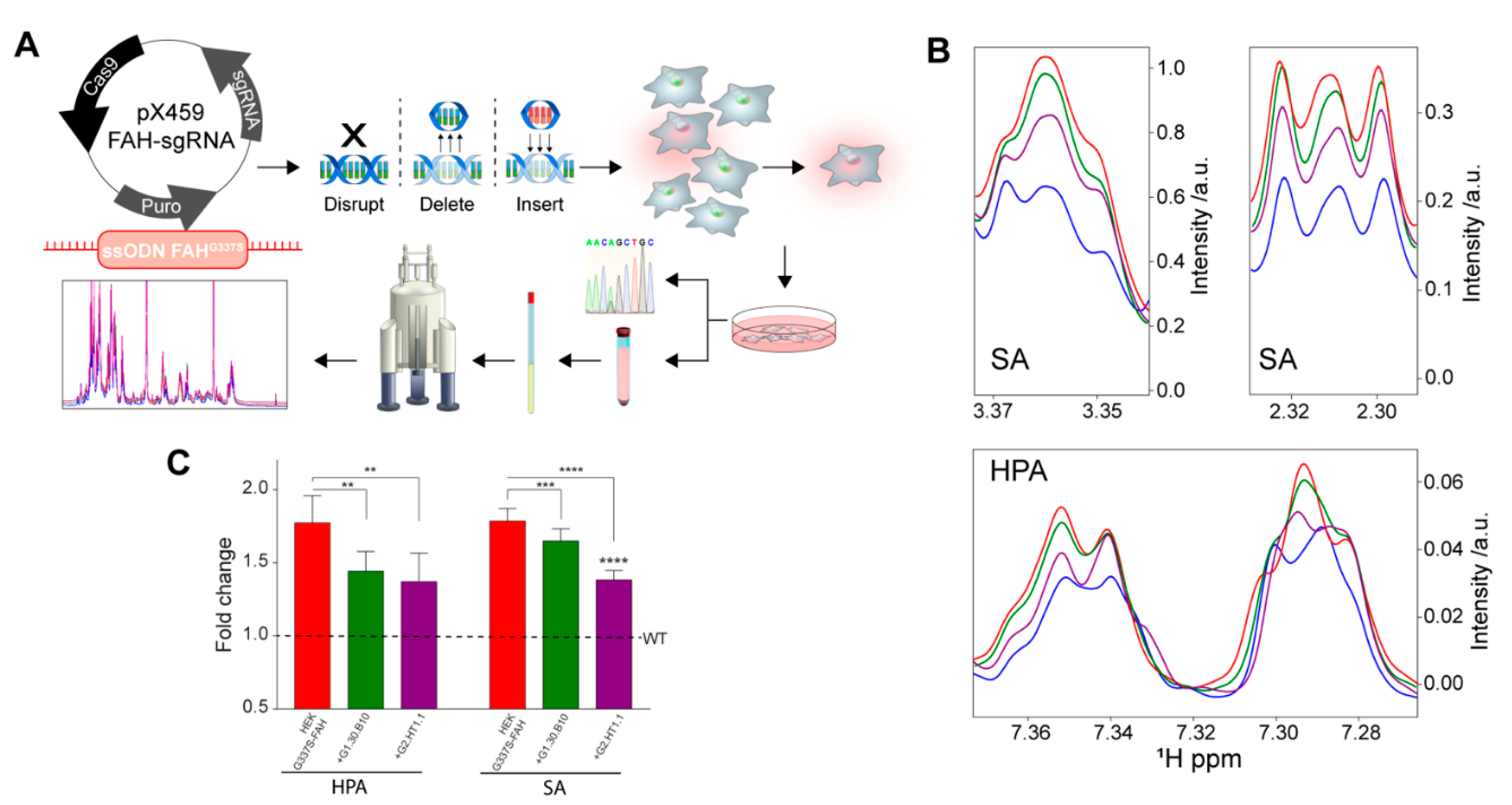
2.7. Inhibitors Restore the Normal Phenotype in a CRISPR/Cas9 Cellular Model of HT1
3. Discussion
4. Materials and Methods
4.1. Computational Analysis
4.2. Protein Production and Purification
4.3. Chemical Libraries
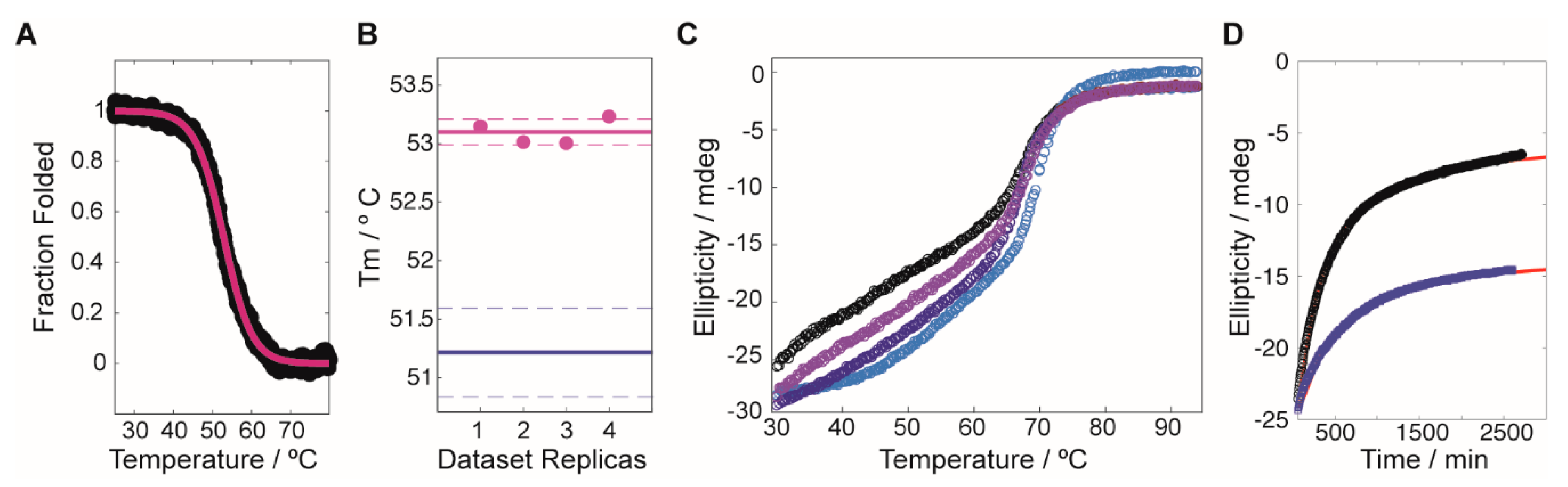
4.4. Protein Thermal Shift Assay
4.5. FAA Synthesis and Purification
4.6. Enzymatic Assay
4.7. NMR Spectroscopy Experiments
4.8. Mammalian Cell Culture and Transfection
4.9. GFP Fluorescence Detection
4.10. Cell Viability Assays (MTT Assay)
4.11. Generation of FAH-G337S Mutant HEK Cells by CRISPR/Cas9-Mediated Genomic Edition
4.12. Metabolite Extraction for NMR
4.13. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Russo, P.A.; Mitchell, G.A.; Tanguay, R.M. Tyrosinemia: A Review. Pediatr. Dev. Pathol. 2001, 4, 212–221. [Google Scholar] [CrossRef]
- Angileri, F.; Bergeron, A.; Morrow, G.; Lettre, F.; Gray, G.; Hutchin, T.; Ball, S.; Tanguay, R.M. Geographical and Ethnic Distribution of Mutations of the Fumarylacetoacetate Hydrolase Gene in Hereditary Tyrosinemia Type 1. In JIMD Rep. 2015, 9, 43–58. [Google Scholar] [CrossRef]
- Tanguay, R.M.; Angileri, F.; Vogel, A. Molecular Pathogenesis of Liver Injury in Hereditary Tyrosinemia 1. In Advances in Experimental Medicine and Biology; Springer Nature: Cham, Switzerland, 2017; pp. 49–64. [Google Scholar]
- Das, A. Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1). Appl. Clin. Genet. 2017, 10, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Geppert, J.; Stinton, C.; Freeman, K.; Fraser, H.; Clarke, A.; Johnson, S.; Sutcliffe, P.; Taylor-Phillips, S. Evaluation of pre-symptomatic nitisinone treatment on long-term outcomes in Tyrosinemia type 1 patients: A systematic review. Orphanet J. Rare Dis. 2017, 12, 154. [Google Scholar] [CrossRef]
- Jack, R.M.; Scott, C.R. Validation of a therapeutic range for nitisinone in patients treated for tyrosinemia type 1 based on reduction of succinylacetone excretion. JIMD Rep. 2019, 46, 75–78. [Google Scholar] [CrossRef]
- Bateman, R.L.; Ashworth, J.; Witte, J.F.; Baker, L.-J.; Bhanumoorthy, P.; Timm, D.E.; Hurley, T.D.; Grompe, M.; Mcclard, R.W. Slow-onset inhibition of fumarylacetoacetate hydrolase by phosphinate mimics of the tetrahedral intermediate: Kinetics, crystal structure and pharmacokinetics. Biochem. J. 2007, 402, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.L.; Bhanumoorthy, P.; Witte, J.F.; McClard, R.W.; Grompe, M.; Timm, D.E. Mechanistic Inferences from the Crystal Structure of Fumarylacetoacetate Hydrolase with a Bound Phosphorus-based Inhibitor. J. Biol. Chem. 2001, 276, 15284–15291. [Google Scholar] [CrossRef] [PubMed]
- Ran, T.; Gao, Y.; Marsh, M.; Zhu, W.; Wang, M.; Mao, X.; Xu, L.; Xu, D.; Wang, W. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. Biochem. J. 2013, 449, 51–60. [Google Scholar] [CrossRef]
- Angileri, F.; Morrow, G.; Roy, V.; Orejuela, D.; Tanguay, R. Heat Shock Response Associated with Hepatocarcinogenesis in a Murine Model of Hereditary Tyrosinemia Type I. Cancers 2014, 6, 998–1019. [Google Scholar] [CrossRef] [PubMed]
- Macias, I.; Laín, A.; Bernardo-Seisdedos, G.; Gil, D.; Gonzalez, E.; Falcon-Perez, J.M.; Millet, O. Hereditary tyrosinemia type I–associated mutations in fumarylacetoacetate hydrolase reduce the enzyme stability and increase its aggregation rate. J. Biol. Chem. 2019, 294, 13051–13060. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Chemical Chaperones: A Pharmacological Strategy for Disorders of Protein Folding and Trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Chemical and pharmacological chaperones as new therapeutic agents. Expert Rev. Mol. Med. 2007, 9, 1–18. [Google Scholar] [CrossRef]
- Urquiza, P.; Laín, A.; Sanz-Parra, A.; Moreno, J.; Bernardo-Seisdedos, G.; Dubus, P.; González, E.; Gutiérrez-de-Juan, V.; García, S.; Eraña, H.; et al. Repurposing ciclopirox as a pharmacological chaperone in a model of congenital erythropoietic porphyria. Sci. Transl. Med. 2018, 10, eaat7467. [Google Scholar] [CrossRef]
- Timm, D.E.; Mueller, H.A.; Bhanumoorthy, P.; Harp, J.M.; Bunick, G.J. Crystal structure and mechanism of a carbon–carbon bond hydrolase. Structure 1999, 7, 1023–1033. [Google Scholar] [CrossRef]
- Pey, A.L.; Ying, M.; Cremades, N.; Velazquez-Campoy, A.; Scherer, T.; Thöny, B.; Sancho, J.; Martinez, A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef]
- Ben Bdira, F.; González, E.; Pluta, P.; Laín, A.; Sanz-Parra, A.; Falcon-Perez, J.M.; Millet, O. Tuning intracellular homeostasis of human uroporphyrinogen III synthase by enzyme engineering at a single hotspot of congenital erythropoietic porphyria. Hum. Mol. Genet. 2014, 23, 5805–5813. [Google Scholar] [CrossRef]
- Fortian, A.; González, E.; Castaño, D.; Falcon-Perez, J.M.; Millet, O. Intracellular Rescue of the Uroporphyrinogen III Synthase Activity in Enzymes Carrying the Hotspot Mutation C73R. J. Biol. Chem. 2011, 286, 13127–13133. [Google Scholar] [CrossRef]
- Viegas, A.; Manso, J.; Corvo, M.C.; Marques, M.M.B.; Cabrita, E.J. Binding of Ibuprofen, Ketorolac, and Diclofenac to COX-1 and COX-2 Studied by Saturation Transfer Difference NMR. J. Med. Chem. 2011, 54, 8555–8562. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, P.; Swan, I.; Nilsson, M.; Morris, G.A. Improving accuracy in DOSY and diffusion measurements using triaxial field gradients. J. Magn. Reson. 2016, 270, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ollerenshaw, J.E.; Tugarinov, V.; Kay, L.E. Methyl TROSY: Explanation and experimental verification. Magn. Reson. Chem. 2003, 41, 843–852. [Google Scholar] [CrossRef]
- Cao, Y.-Y.; Zhang, Y.-L.; DU, J.; Qu, Y.-J.; Zhong, X.-M.; Bai, J.-L.; Song, F. Compound mutations (R237X and L375P) in the fumarylacetoacetate hydrolase gene causing tyrosinemia type I in a Chinese patient. Chin. Med. J. (Engl.) 2012, 125, 2132–2136. [Google Scholar] [PubMed]
- Aktuglu Zeybek, A.C.; Kiykim, E.; Soyucen, E.; Cansever, S.; Altay, S.; Zubarioglu, T.; Erkan, T.; Aydin, A. Hereditary tyrosinemia type 1 in Turkey: Twenty year single-center experience. Pediatr. Int. 2015, 57, 281–289. [Google Scholar] [CrossRef]
- Couce, M.L.; Dalmau, J.; del Toro, M.; Pintos-Morell, G.; Aldámiz-Echevarría, L. Tyrosinemia type 1 in Spain: Mutational analysis, treatment and long-term outcome. Pediatr. Int. 2011, 53, 985–989. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Mathews, M.A.A. Crystal structure of human uroporphyrinogen III synthase. EMBO J. 2001, 20, 5832–5839. [Google Scholar] [CrossRef]
- Bernardo-Seisdedos, G.; Nuñez, E.; Gomis-Perez, C.; Malo, C.; Villarroel, Á.; Millet, O. Structural basis and energy landscape for the Ca 2+ gating and calmodulation of the Kv7.2 K + channel. Proc. Natl. Acad. Sci. USA 2018, 115, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cañón, J.M.; Peñalva, M.A. Spectrophotometric Determination of Homogentisate UsingAspergillus nidulansHomogentisate Dioxygenase. Anal. Biochem. 1997, 245, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Jerschow, A.; Müller, N. Efficient Simulation of Coherence Transfer Pathway Selection by Phase Cycling and Pulsed Field Gradients in NMR. J. Magn. Reson. 1998, 134, 17–29. [Google Scholar] [CrossRef][Green Version]
- Anderson, R.C.; Lin, M.; Shapiro, M.J. Affinity NMR: Decoding DNA Binding. J. Comb. Chem. 1999, 1, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Millet, O.; Bernadó, P.; Garcia, J.; Rizo, J.; Pons, M. NMR measurement of the off rate from the first calcium-binding site of the synaptotagmin I C 2 A domain. FEBS Lett. 2002, 516, 93–96. [Google Scholar] [CrossRef]
- Nevozhay, D. Cheburator software for automatically calculating drug inhibitory concentrations from in vitroscreening assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil-Martínez, J.; Macias, I.; Unione, L.; Bernardo-Seisdedos, G.; Lopitz-Otsoa, F.; Fernandez-Ramos, D.; Lain, A.; Sanz-Parra, A.; Mato, J.M.; Millet, O. Therapeutic Targeting of Fumaryl Acetoacetate Hydrolase in Hereditary Tyrosinemia Type I. Int. J. Mol. Sci. 2021, 22, 1789. https://doi.org/10.3390/ijms22041789
Gil-Martínez J, Macias I, Unione L, Bernardo-Seisdedos G, Lopitz-Otsoa F, Fernandez-Ramos D, Lain A, Sanz-Parra A, Mato JM, Millet O. Therapeutic Targeting of Fumaryl Acetoacetate Hydrolase in Hereditary Tyrosinemia Type I. International Journal of Molecular Sciences. 2021; 22(4):1789. https://doi.org/10.3390/ijms22041789
Chicago/Turabian StyleGil-Martínez, Jon, Iratxe Macias, Luca Unione, Ganeko Bernardo-Seisdedos, Fernando Lopitz-Otsoa, David Fernandez-Ramos, Ana Lain, Arantza Sanz-Parra, José M Mato, and Oscar Millet. 2021. "Therapeutic Targeting of Fumaryl Acetoacetate Hydrolase in Hereditary Tyrosinemia Type I" International Journal of Molecular Sciences 22, no. 4: 1789. https://doi.org/10.3390/ijms22041789
APA StyleGil-Martínez, J., Macias, I., Unione, L., Bernardo-Seisdedos, G., Lopitz-Otsoa, F., Fernandez-Ramos, D., Lain, A., Sanz-Parra, A., Mato, J. M., & Millet, O. (2021). Therapeutic Targeting of Fumaryl Acetoacetate Hydrolase in Hereditary Tyrosinemia Type I. International Journal of Molecular Sciences, 22(4), 1789. https://doi.org/10.3390/ijms22041789