
Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery



Abstract
1. Introduction
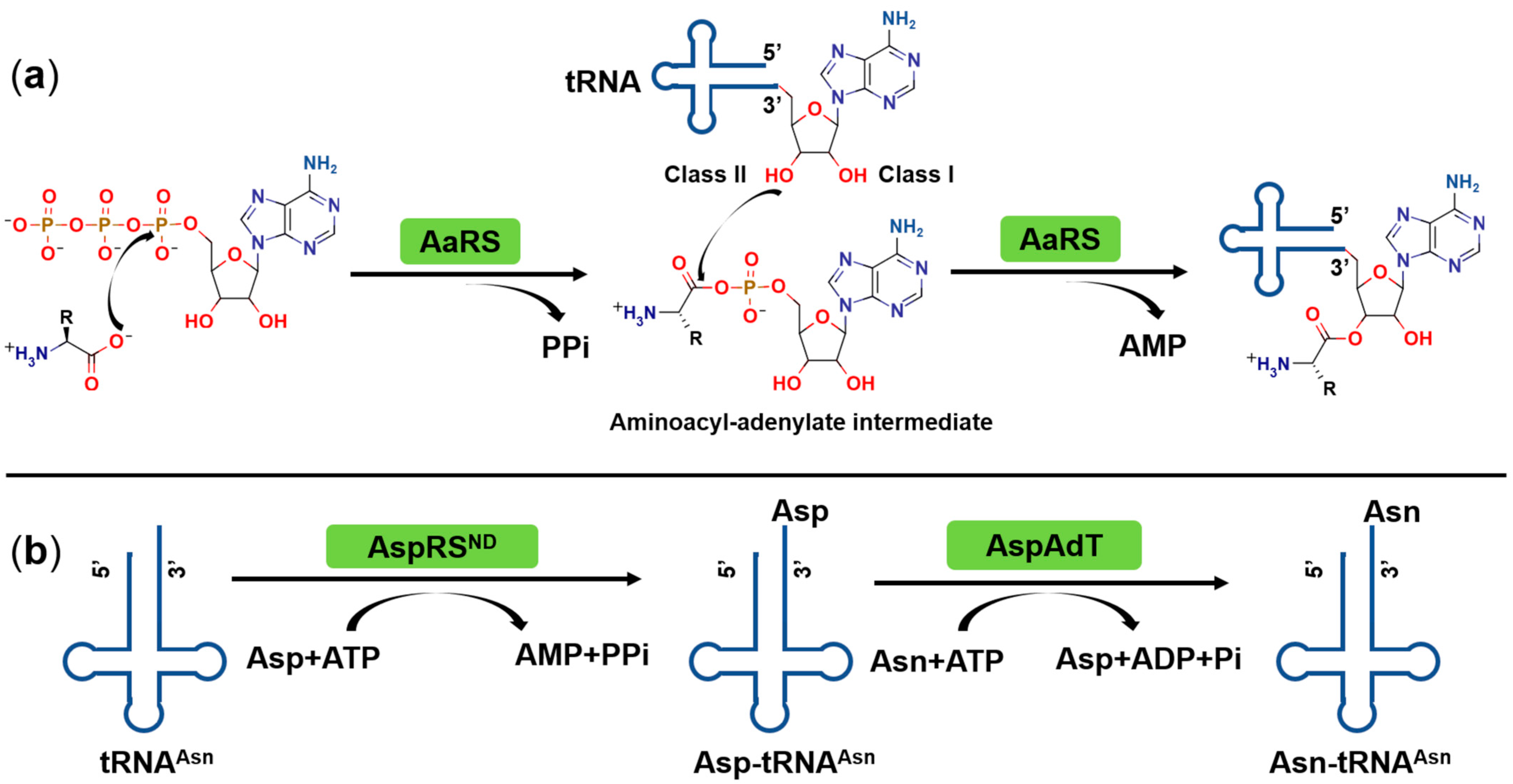
2. Catalytic Mechanism of Aminoacyl-tRNA Synthetases (aaRSs)
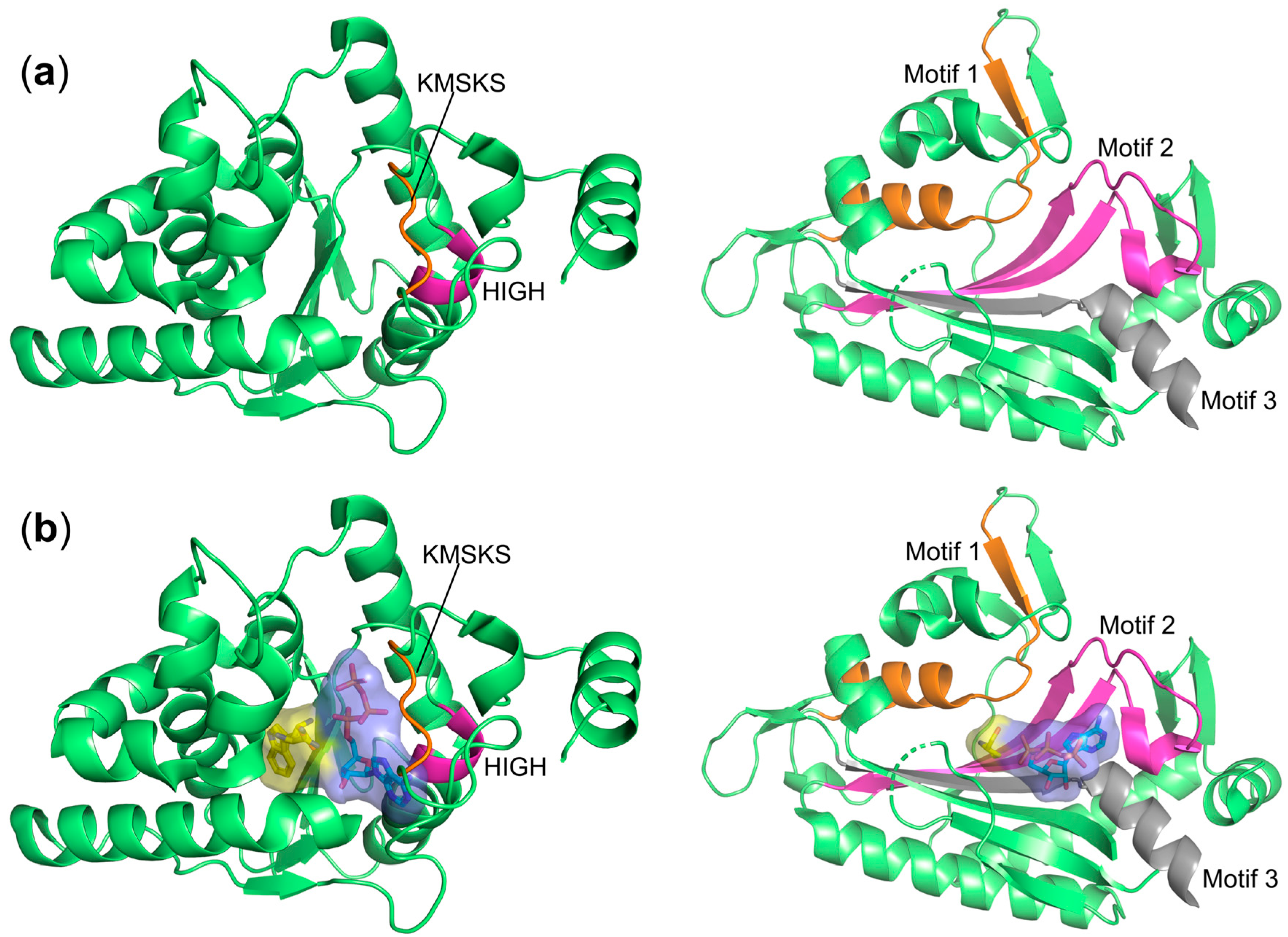
3. Classification of aaRSs
4. Quality Control for Correct Loading of aaRSs
5. Why Are aaRSs Valuable Targets for Anti-Infective Drug Discovery?
6. Natural and Synthetic aaRSs Inhibitors and Their Inhibitory Mechanisms
6.1. Targeting the Active Site
6.1.1. Targeting the Amino Acid Binding Site
6.1.2. Targeting the ATP Binding Site
6.1.3. Targeting the Amino Acid and 3′-End of tRNA Bi-Substrate Binding Site
6.1.4. Targeting the ATP and 3′-End of tRNA Bi-Substrate Binding Site
6.1.5. Targeting Both the Amino Acid and an Auxiliary Site
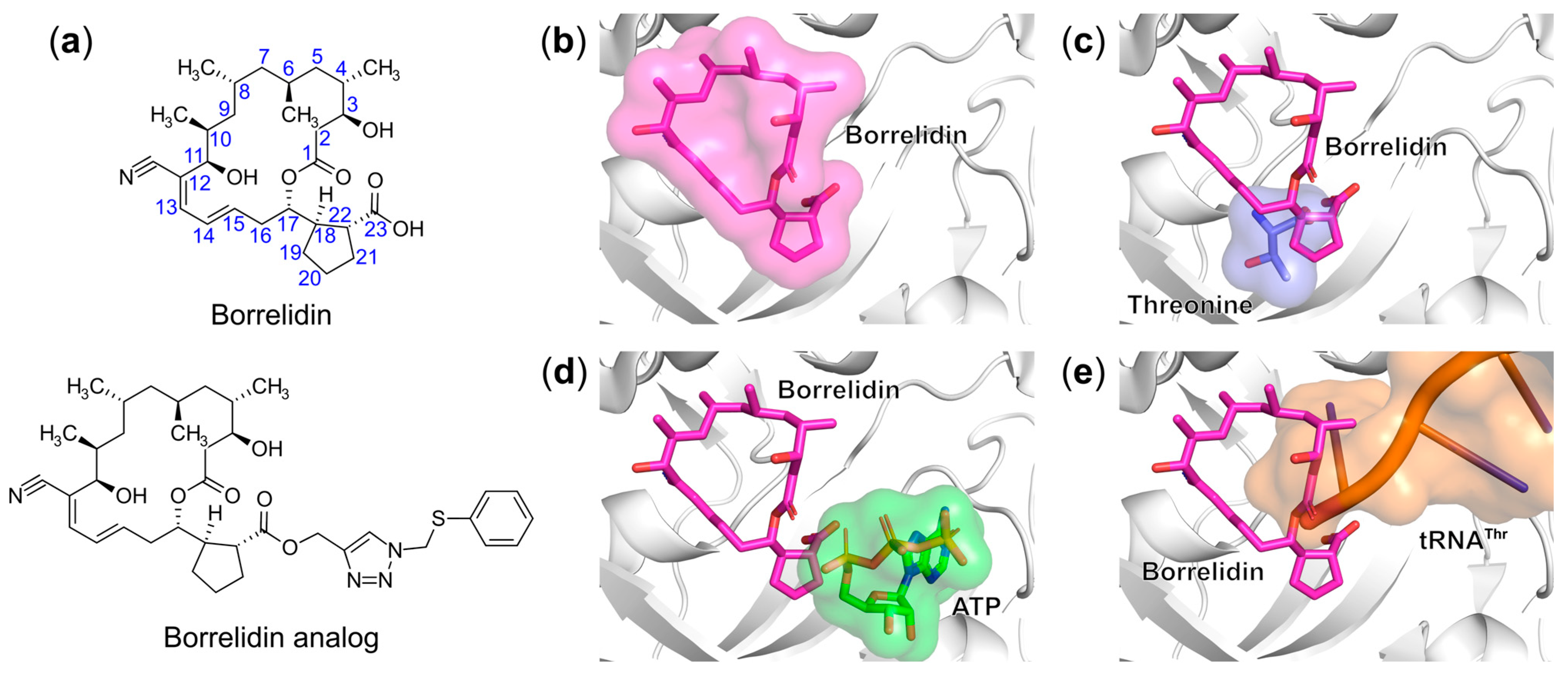
6.1.6. Targeting the Aminoacyl-Adenylate Bi-Substrate Binding Site
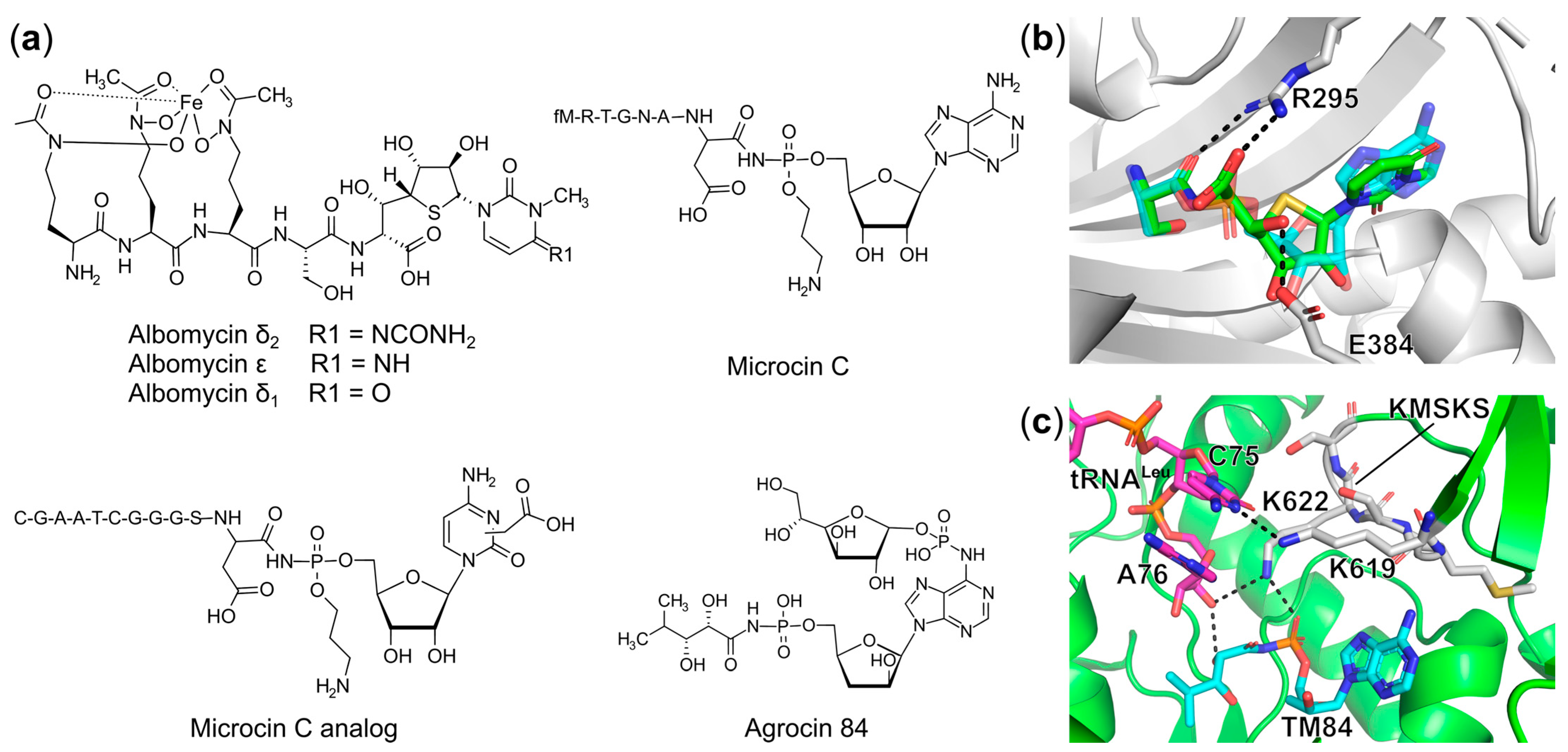
Trojan Horse aaRS Inhibitors
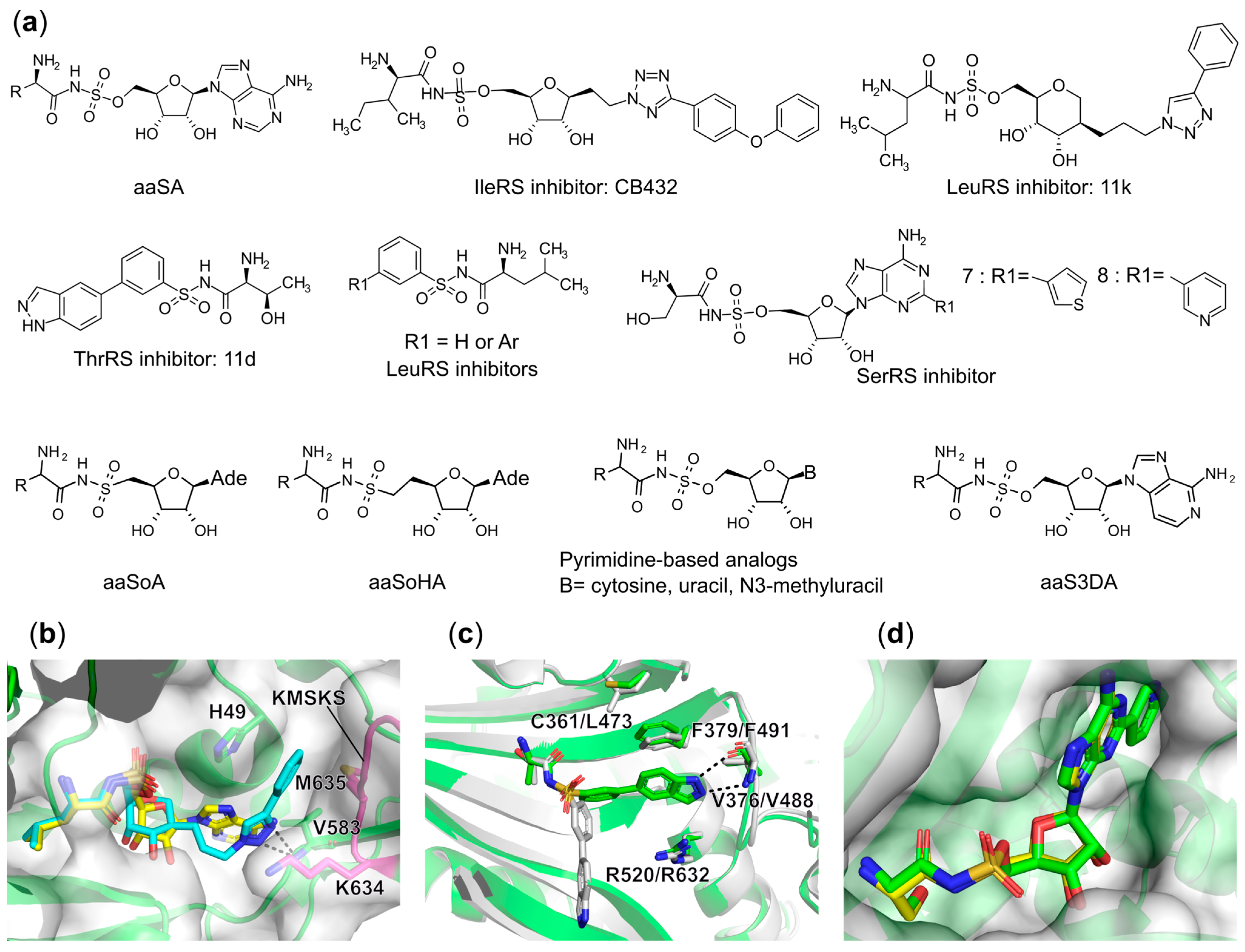
Synthetic Bi-Substrate aaRS Inhibitors
6.1.7. Targeting Multiple Binding Sites
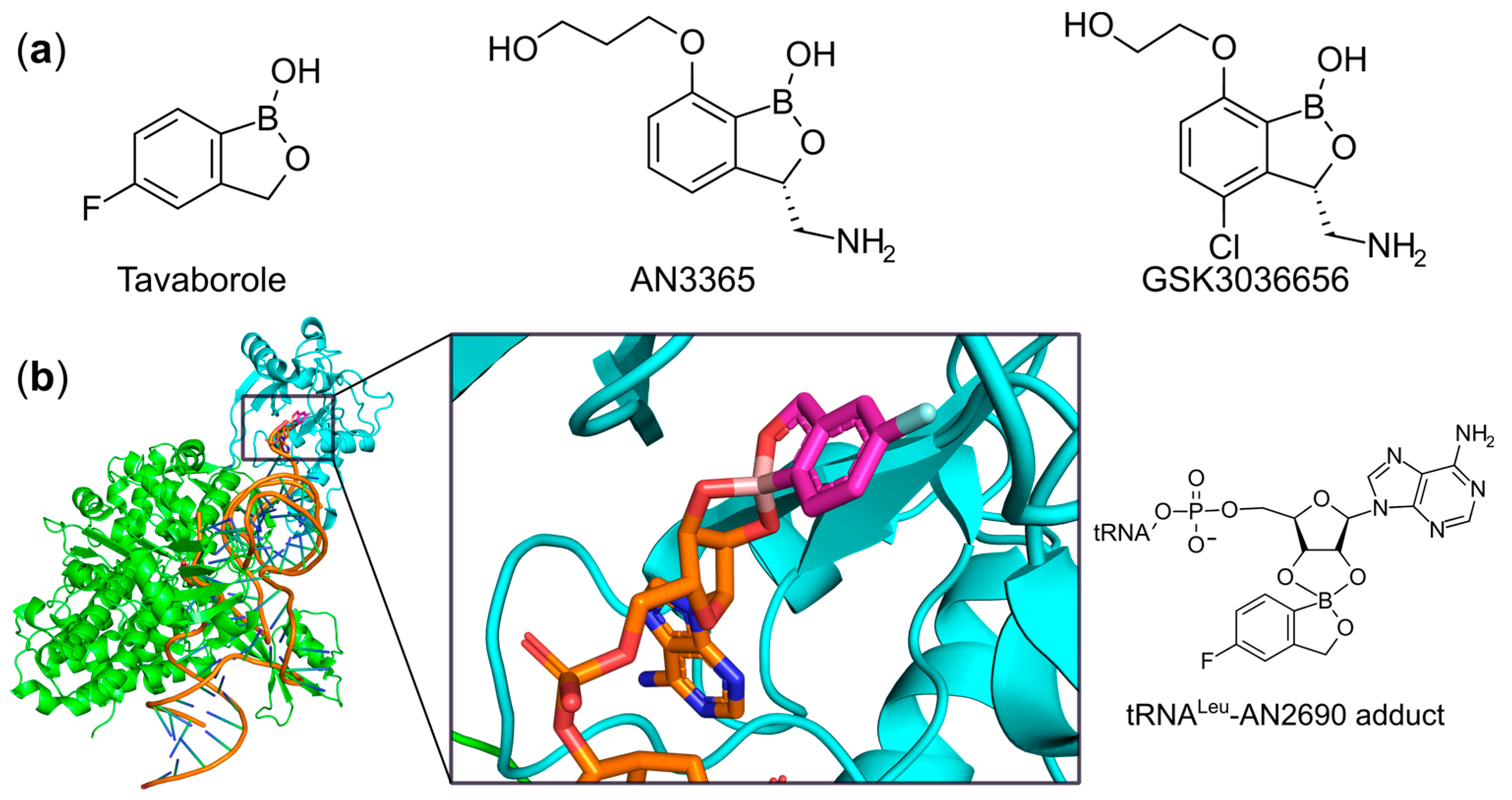
6.2. Targeting the Editing Site
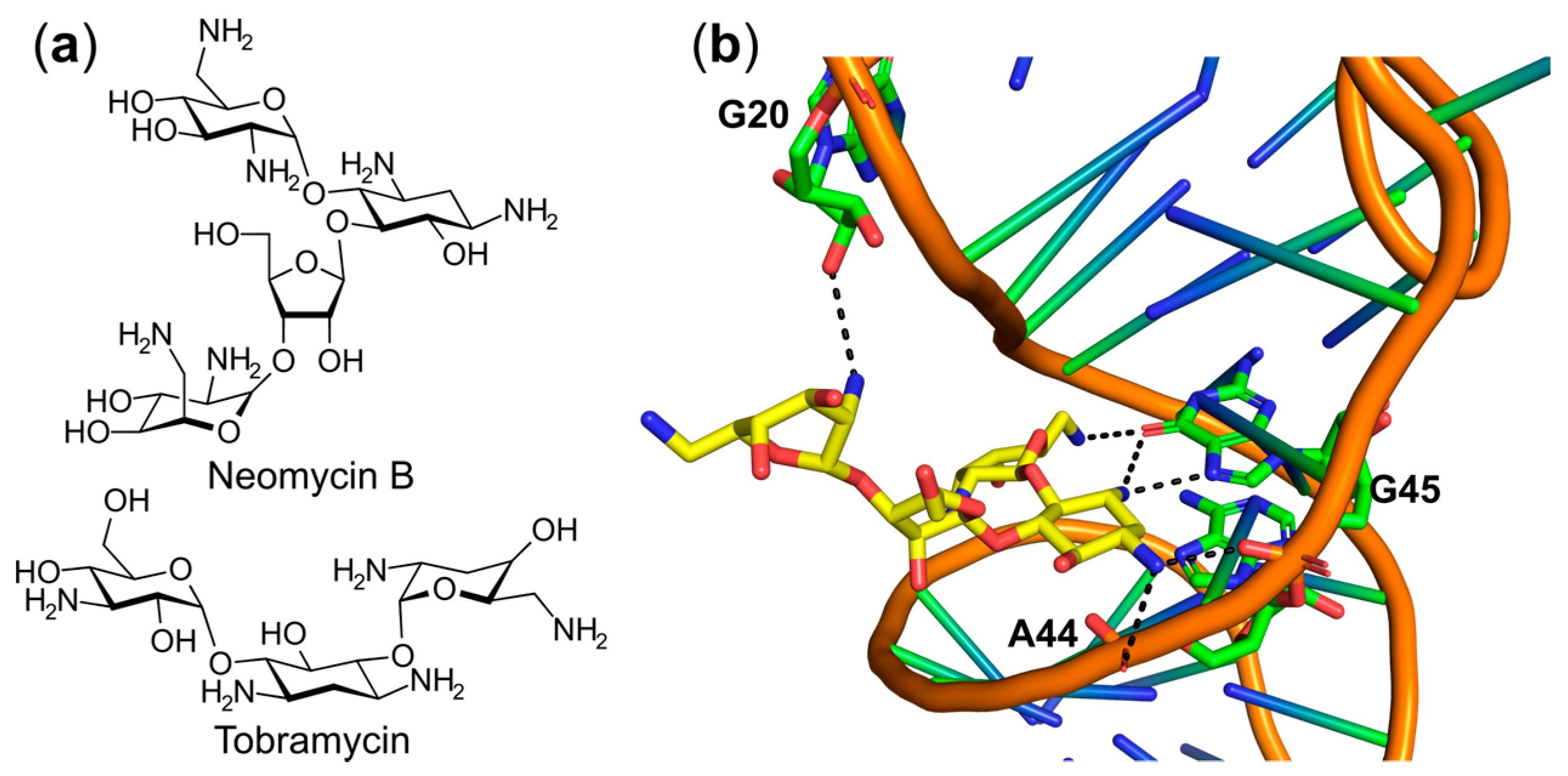
6.3. Targeting the tRNA or the aaRS–tRNA Interface
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa-AMP | aminoacyl-adenylate |
| aaRS | aminoacyl-tRNA synthetase |
| aa-tRNA | aminoacyl-tRNA |
| aaS3DA | aminoacyl sulfamoyl 3-deazaadenosine |
| aaSA | aminoacyl-sulfamoyl adenosine |
| aaSoA | 5′-(N-aminoacyl)-sulfonamido-5′-deoxyadenosine |
| aaSoHA | N-L-aminoacyl-C-5′-adenosyl-methansulfonamide |
| AdT | amidotransferase |
| AlaRS | alanyl-tRNA synthetase |
| AMP | adenosine monophosphate |
| AMPPNP | adenylyl-imidodiphosphate |
| AMR | antimicrobial resistance |
| AP | auxiliary pocket |
| ArgRS | arginyl-tRNA synthetase |
| AsnRS | asparaginyl-tRNA synthetase |
| AspRS | aspartyl-tRNA synthetase |
| ATP | adenosine triphosphate |
| CP1 | connective polypeptide 1 |
| CysRS | cysteinyl-RNA synthetase |
| DNA | Deoxyribonucleic acid |
| EC50 | half maximal effective concentration |
| EPRS | glutamyl prolyl-tRNA synthetase |
| ESBL | extended spectrum β-lactamase |
| FDA | Food and Drug Administration |
| GlnRS | glutaminyl-tRNA synthetase |
| GluRS | glutamyl-tRNA synthetase |
| GlyRS | glycyl-tRNA synthetase |
| GSA | glycyl-sulfamoyl adenosine |
| GSK | GlaxoSmithKline |
| HisRS | histidyl-tRNA synthetase |
| HTS | high-throughput screening |
| IC50 | half maximal inhibitory concentration |
| IleRS | isoleucyl-tRNA synthetase |
| ISA | isoleucyl-sulfamoyl adenosine |
| ITC | isothermal titration calorimetry |
| LeuRS | leucyl-tRNA synthetase |
| LSA | leucyl-sulfamoyl adenosine |
| LysRS | lysyl-tRNA synthetase |
| McC | microcin C |
| MDR | multidrug resistance |
| MetRS | methionyl-tRNA synthetase |
| MIC | minimum inhibitory concentration |
| MRSA | methicillin-resistant Staphylococcus aureus |
| Mtb | Mycobacterium tuberculosis |
| PDB | protein data bank |
| PheRS | phenylalanyl-tRNA synthetase |
| PPi | pyrophosphate |
| ProRS | prolyl-tRNA synthetase |
| PylRS | pyrrolysyl-tRNA synthetase |
| RF | Rossmann fold |
| RNA | ribonucleic acid |
| SAR | Structure-activity relationship |
| SBDD | structure-based drug design |
| SepRS | phosphoseryl-tRNA synthetase |
| SerRS | seryl-tRNA synthetase |
| SSA | seryl-sulfamoyl adenosine |
| ThrRS | threonyl-tRNA synthetase |
| Tm | melting temperature |
| tRNA | transfer RNA |
| TrpRS | tryptophanyl-tRNA synthetase |
| TSA | threonyl-sulfamoyl adenosine |
| TyrRS | tyrosyl-tRNA synthetase |
| ValRS | valyl-tRNA synthetase |
| WHO | World Health Organization |
References
- Infectious Diseases Society of America (IDSA). Combating Antimicrobial Resistance: Policy Recommendations to Save Lives. Clin. Infect. Dis. 2011, 52, S397–S428. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- WHO. Antimicrobial Resistance: Global Report on Surveillance 2014. Available online: http://www.who.int/antimicrobial-resistance/publications/surveillancereport/en/ (accessed on 4 April 2020).
- Lewis, K. Platforms for Antibiotic Discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L. General Principles of Antibiotic Resistance in Bacteria. Drug Discov. Today Technol. 2014, 11, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Publications. AMR Review. Available online: https://amr-review.org/Publications.html (accessed on 4 April 2020).
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 11 January 2021).
- Ling, J.; Reynolds, N.; Ibba, M. Aminoacyl-TRNA Synthesis and Translational Quality Control. Annu. Rev. Microbiol. 2009, 63, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Soll, D. Aminoacyl-TRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W. Coding of Class I and II Aminoacyl-TRNA Synthetases. Adv. Exp. Med. Biol. 2017, 966, 103–148. [Google Scholar] [CrossRef]
- Yao, P.; Fox, P.L. Aminoacyl-TRNA Synthetases in Medicine and Disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef]
- Guo, M.; Schimmel, P. Essential Non-Translational Functions of TRNA Synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Kim, S.; Kim, M.H. Aminoacyl-TRNA Synthetases, Therapeutic Targets for Infectious Diseases. Biochem. Pharmacol. 2018, 154, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.W. What is an Antibiotic? In Antibiotics: Current Innovations and Future Trends; Caister Academic Press: Poole, UK, 2015; pp. 1–18. ISBN 978-1-908230-54-6. [Google Scholar]
- Ibba, M.; Söll, D. The Renaissance of Aminoacyl-TRNA Synthesis. EMBO Rep. 2001, 2, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Ravel, J.M.; Wang, S.F.; Heinemeyer, C.; Shive, W. Glutamyl and Glutaminyl Ribonucleic Acid Synthetases of Escherichia Coli W. Separation, Properties, and Stimulation of Adenosine Triphosphate-Pyrophosphate Exchange by Acceptor Ribonucleic Acid. J. Biol. Chem. 1965, 240, 432–438. [Google Scholar] [CrossRef]
- Ibba, M.; Losey, H.C.; Kawarabayasi, Y.; Kikuchi, H.; Bunjun, S.; Söll, D. Substrate Recognition by Class I Lysyl-TRNA Synthetases: A Molecular Basis for Gene Displacement. Proc. Natl. Acad. Sci. USA 1999, 96, 418–423. [Google Scholar] [CrossRef]
- Kern, D.; Lapointe, J. Glutamyl Transfer Ribonucleic Acid Synthetase of Escherichia Coli. Study of the Interactions with Its Substrates. Biochemistry 1979, 18, 5809–5818. [Google Scholar] [CrossRef]
- Mehler, A.H.; Mitra, S.K. The Activation of Arginyl Transfer Ribonucleic Acid Synthetase by Transfer Ribonucleic Acid. J. Biol. Chem. 1967, 242, 5495–5499. [Google Scholar] [CrossRef]
- Sheppard, K.; Yuan, J.; Hohn, M.J.; Jester, B.; Devine, K.M.; Söll, D. From One Amino Acid to Another: TRNA-Dependent Amino Acid Biosynthesis. Nucleic Acids Res. 2008, 36, 1813–1825. [Google Scholar] [CrossRef]
- Tumbula, D.L.; Becker, H.D.; Chang, W.Z.; Söll, D. Domain-Specific Recruitment of Amide Amino Acids for Protein Synthesis. Nature 2000, 407, 106–110. [Google Scholar] [CrossRef]
- Gagnon, Y.; Lacoste, L.; Champagne, N.; Lapointe, J. Widespread Use of the Glu-TRNAGln Transamidation Pathway among Bacteria. A Member of the Alpha Purple Bacteria Lacks Glutaminyl-Trna Synthetase. J. Biol. Chem. 1996, 271, 14856–14863. [Google Scholar] [CrossRef]
- Curnow, A.W.; Hong, K.W.; Yuan, R.; Kim, S.I.; Martins, O.; Winkler, W.; Henkin, T.M.; Söll, D. Glu-TRNAGln Amidotransferase: A Novel Heterotrimeric Enzyme Required for Correct Decoding of Glutamine Codons during Translation. Proc. Natl. Acad. Sci. USA 1997, 94, 11819–11826. [Google Scholar] [CrossRef] [PubMed]
- Kamtekar, S.; Hohn, M.J.; Park, H.-S.; Schnitzbauer, M.; Sauerwald, A.; Söll, D.; Steitz, T.A. Toward Understanding Phosphoseryl-TRNACys Formation: The Crystal Structure of Methanococcus Maripaludis Phosphoseryl-TRNA Synthetase. Proc. Natl. Acad. Sci. USA 2007, 104, 2620–2625. [Google Scholar] [CrossRef]
- Gaston, M.A.; Jiang, R.; Krzycki, J.A. Functional Context, Biosynthesis, and Genetic Encoding of Pyrrolysine. Curr. Opin. Microbiol. 2011, 14, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Sauerwald, A.; Zhu, W.; Major, T.A.; Roy, H.; Palioura, S.; Jahn, D.; Whitman, W.B.; Yates, J.R.; Ibba, M.; Söll, D. RNA-Dependent Cysteine Biosynthesis in Archaea. Science 2005, 307, 1969–1972. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Fang, X.; Wallace, A.L.; Wu, B.; Liu, W.R. A Rationally Designed Pyrrolysyl-TRNA Synthetase Mutant with a Broad Substrate Spectrum. J. Am. Chem. Soc. 2012, 134, 2950–2953. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-S.; Fang, X.; Chen, H.-Y.; Wu, B.; Wang, Z.U.; Hilty, C.; Liu, W.R. Genetic Incorporation of Twelve Meta-Substituted Phenylalanine Derivatives Using a Single Pyrrolysyl-TRNA Synthetase Mutant. ACS Chem. Biol. 2013, 8, 405–415. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, H.; Chen, Y.; Zhang, B.; Yang, Y.; Zang, J.; Wu, J.; Lin, S. Chimeric Design of Pyrrolysyl-TRNA Synthetase/TRNA Pairs and Canonical Synthetase/TRNA Pairs for Genetic Code Expansion. Nat. Commun. 2020, 11, 3154. [Google Scholar] [CrossRef] [PubMed]
- Antonellis, A.; Green, E.D. The Role of Aminoacyl-TRNA Synthetases in Genetic Diseases. Annu Rev. Genom. Hum. Genet. 2008, 9, 87–107. [Google Scholar] [CrossRef]
- Martin, W.F.; Garg, S.; Zimorski, V. Endosymbiotic Theories for Eukaryote Origin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Brick, P.; Bhat, T.N.; Blow, D.M. Structure of Tyrosyl-TRNA Synthetase Refined at 2.3 Å Resolution: Interaction of the Enzyme with the Tyrosyl Adenylate Intermediate. J. Mol. Biol. 1989, 208, 83–98. [Google Scholar] [CrossRef]
- Zelwer, C.; Risler, J.L.; Brunie, S. Crystal Structure of Escherichia Coli Methionyl-TRNA Synthetase at 2.5 Å Resolution. J. Mol. Biol. 1982, 155, 63–81. [Google Scholar] [CrossRef]
- Rould, M.A.; Perona, J.J.; Söll, D.; Steitz, T.A. Structure of E. Coli Glutaminyl-TRNA Synthetase Complexed with TRNA(Gln) and ATP at 2.8 A Resolution. Science 1989, 246, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W. Cognition, Mechanism, and Evolutionary Relationships in Aminoacyl-TRNA Synthetases. Annu. Rev. Biochem. 1993, 62, 715–748. [Google Scholar] [CrossRef]
- Perona, J.J.; Hadd, A. Structural Diversity and Protein Engineering of the Aminoacyl-TRNA Synthetases. Biochemistry 2012, 51, 8705–8729. [Google Scholar] [CrossRef]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of TRNA Synthetases into Two Classes Based on Mutually Exclusive Sets of Sequence Motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef]
- Cusack, S.; Berthet-Colominas, C.; Härtlein, M.; Nassar, N.; Leberman, R. A Second Class of Synthetase Structure Revealed by X-ray Analysis of Escherichia Coli Seryl-TRNA Synthetase at 2.5 A. Nature 1990, 347, 249–255. [Google Scholar] [CrossRef]
- Ruff, M.; Krishnaswamy, S.; Boeglin, M.; Poterszman, A.; Mitschler, A.; Podjarny, A.; Rees, B.; Thierry, J.C.; Moras, D. Class II Aminoacyl Transfer RNA Synthetases: Crystal Structure of Yeast Aspartyl-TRNA Synthetase Complexed with TRNA(Asp). Science 1991, 252, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Kalita, P.; Shukla, H.; Kumar, A.; Tripathi, T. Aminoacyl-TRNA Synthetases: Structure, Function, and Drug Discovery. Int. J. Biol. Macromol. 2018, 111, 400–414. [Google Scholar] [CrossRef]
- Sprinzl, M. Chemistry of Aminoacylation and Peptide Bond Formation on the 3′terminus of TRNA. J. Biosci. 2006, 31, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P. Development of TRNA Synthetases and Connection to Genetic Code and Disease. Protein Sci. 2008, 17, 1643–1652. [Google Scholar] [CrossRef]
- O’Donoghue, P.; Luthey-Schulten, Z. On the Evolution of Structure in Aminoacyl-TRNA Synthetases. Microbiol. Mol. Biol. Rev. 2003, 67, 550–573. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S. Eleven down and Nine to Go. Nat. Struct. Biol. 1995, 2, 824–831. [Google Scholar] [CrossRef]
- Delarue, M.; Moras, D. The Aminoacyl-TRNA Synthetase Family: Modules at Work. Bioessays 1993, 15, 675–687. [Google Scholar] [CrossRef]
- Yadavalli, S.S.; Ibba, M. Quality Control in Aminoacyl-TRNA Synthesis Its Role in Translational Fidelity. Adv. Protein Chem. Struct. Biol. 2012, 86, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Karkhanis, V.A.; Mascarenhas, A.P.; Martinis, S.A. Amino Acid Toxicities of Escherichia Coli That Are Prevented by Leucyl-TRNA Synthetase Amino Acid Editing. J. Bacteriol. 2007, 189, 8765–8768. [Google Scholar] [CrossRef]
- Lee, J.W.; Beebe, K.; Nangle, L.A.; Jang, J.; Longo-Guess, C.M.; Cook, S.A.; Davisson, M.T.; Sundberg, J.P.; Schimmel, P.; Ackerman, S.L. Editing-Defective TRNA Synthetase Causes Protein Misfolding and Neurodegeneration. Nature 2006, 443, 50–55. [Google Scholar] [CrossRef]
- Tian, Q.; Wang, C.; Liu, Y.; Xie, W. Structural Basis for Recognition of G-1-Containing TRNA by Histidyl-TRNA Synthetase. Nucleic Acids Res. 2015, 43, 2980–2990. [Google Scholar] [CrossRef]
- Hou, Y.M.; Westhof, E.; Giegé, R. An Unusual RNA Tertiary Interaction Has a Role for the Specific Aminoacylation of a Transfer RNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6776–6780. [Google Scholar] [CrossRef]
- Naganuma, M.; Sekine, S.; Chong, Y.E.; Guo, M.; Yang, X.-L.; Gamper, H.; Hou, Y.-M.; Schimmel, P.; Yokoyama, S. The Selective TRNA Aminoacylation Mechanism Based on a Single G•U Pair. Nature 2014, 510, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S.; Gruic-Sovulj, I. How Evolution Shapes Enzyme Selectivity—Lessons from Aminoacyl-TRNA Synthetases and Other Amino Acid Utilizing Enzymes. FEBS J. 2020, 287, 1284–1305. [Google Scholar] [CrossRef]
- Fersht, A.R.; Kaethner, M.M. Enzyme Hyperspecificity. Rejection of Threonine by the Valyl-TRNA Synthetase by Misacylation and Hydrolytic Editing. Biochemistry 1976, 15, 3342–3346. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. Editing Mechanisms in Protein Synthesis. Rejection of Valine by the Isoleucyl-TRNA Synthetase. Biochemistry 1977, 16, 1025–1030. [Google Scholar] [CrossRef]
- Gruic-Sovulj, I.; Rokov-Plavec, J.; Weygand-Durasevic, I. Hydrolysis of Non-Cognate Aminoacyl-Adenylates by a Class II Aminoacyl-TRNA Synthetase Lacking an Editing Domain. FEBS Lett. 2007, 581, 5110–5114. [Google Scholar] [CrossRef] [PubMed]
- Yadavalli, S.S.; Musier-Forsyth, K.; Ibba, M. The Return of Pretransfer Editing in Protein Synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 19031–19032. [Google Scholar] [CrossRef] [PubMed]
- Palencia, A.; Crépin, T.; Vu, M.T.; Lincecum, T.L.; Martinis, S.A.; Cusack, S. Structural Dynamics of the Aminoacylation and Proofreading Functional Cycle of Bacterial Leucyl-TRNA Synthetase. Nat. Struct. Mol. Biol. 2012, 19, 677–684. [Google Scholar] [CrossRef]
- Zhang, C.-M.; Perona, J.J.; Ryu, K.; Francklyn, C.; Hou, Y.-M. Distinct Kinetic Mechanisms of the Two Classes of Aminoacyl-TRNA Synthetases. J. Mol. Biol. 2006, 361, 300–311. [Google Scholar] [CrossRef]
- Beebe, K.; Mock, M.; Merriman, E.; Schimmel, P. Distinct Domains of TRNA Synthetase Recognize the Same Base Pair. Nature 2008, 451, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Kuzmishin Nagy, A.B.; Bakhtina, M.; Musier-Forsyth, K. Chapter Four—Trans-editing by aminoacyl-tRNA synthetase-like editing domains. In The Enzymes; Biology of Aminoacyl-tRNA Synthetases; Ribas de Pouplana, L., Kaguni, L.S., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 48, pp. 69–115. [Google Scholar]
- An, S.; Musier-Forsyth, K. Trans-Editing of Cys-TRNAPro by Haemophilus Influenzae YbaK Protein. J. Biol. Chem. 2004, 279, 42359–42362. [Google Scholar] [CrossRef]
- Korencic, D.; Ahel, I.; Schelert, J.; Sacher, M.; Ruan, B.; Stathopoulos, C.; Blum, P.; Ibba, M.; Söll, D. A Freestanding Proofreading Domain Is Required for Protein Synthesis Quality Control in Archaea. Proc. Natl. Acad. Sci. USA 2004, 101, 10260–10265. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Chong, Y.E.; Beebe, K.; Shapiro, R.; Yang, X.-L.; Schimmel, P. The C-Ala Domain Brings Together Editing and Aminoacylation Functions on One TRNA. Science 2009, 325, 744–747. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How Antibiotics Kill Bacteria: From Targets to Networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Sun, X.; Jarvis, T.; Critchley, I.; Janjic, N. Aminoacyl-TRNA Synthetases: Essential and Still Promising Targets for New Anti-Infective Agents. Expert Opin. Investig. Drugs 2007, 16, 573–593. [Google Scholar] [CrossRef]
- Francklyn, C.S.; Mullen, P. Progress and Challenges in Aminoacyl-TRNA Synthetase-Based Therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef]
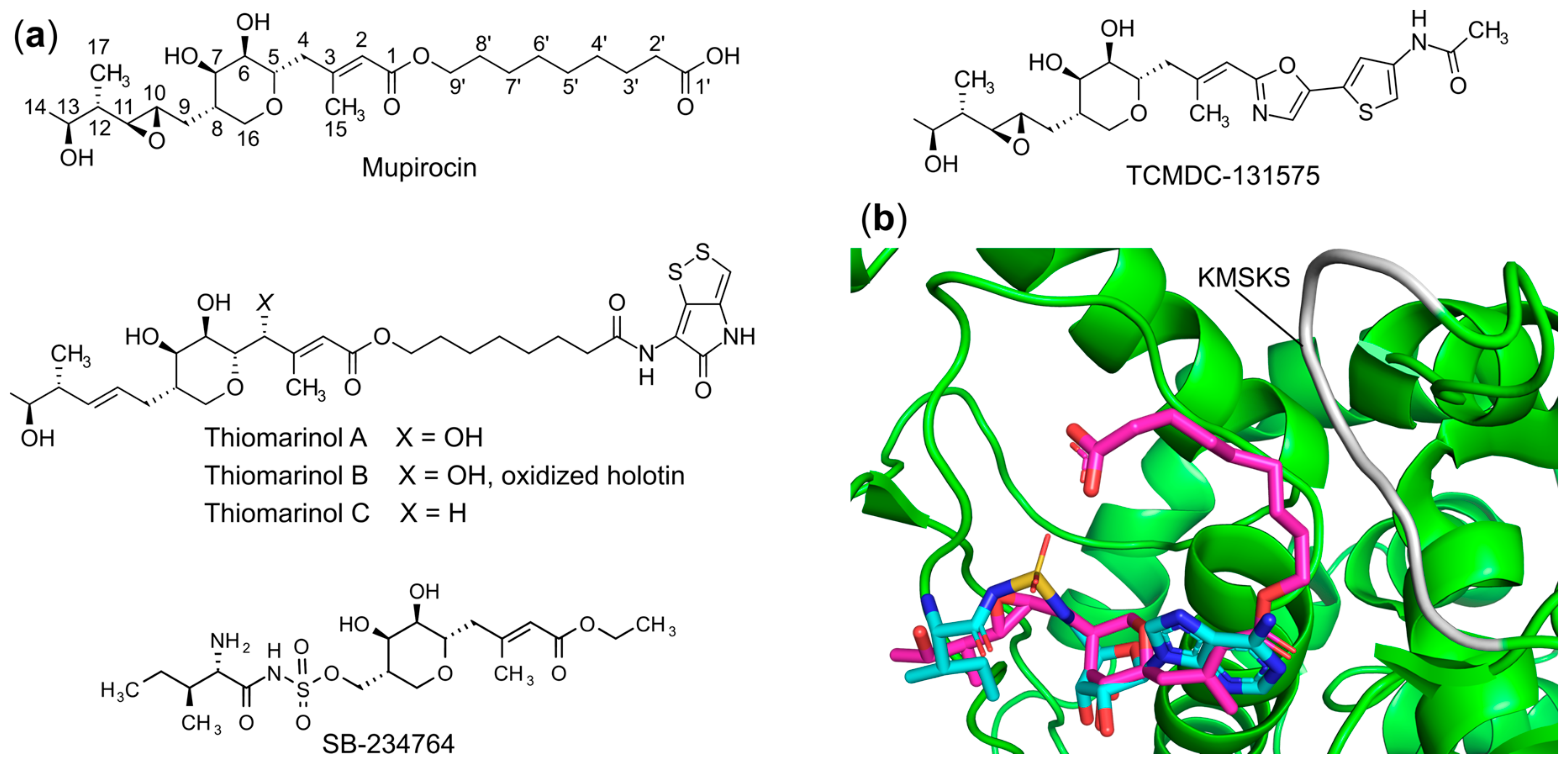
- Nakama, T.; Nureki, O.; Yokoyama, S. Structural Basis for the Recognition of Isoleucyl-Adenylate and an Antibiotic, Mupirocin, by Isoleucyl-TRNA Synthetase. J. Biol. Chem. 2001, 276, 47387–47393. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crépin, T.; Zhou, H.; Zhang, Y.-K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An Antifungal Agent Inhibits an Aminoacyl-TRNA Synthetase by Trapping TRNA in the Editing Site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Vondenhoff, G.H.M.; Van Aerschot, A. Aminoacyl-TRNA Synthetase Inhibitors as Potential Antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P.; Tao, J.; Hill, J. Aminoacyl TRNA Synthetases as Targets for New Anti-Infectives. FASEB J. 1998, 12, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Kwon, N.H.; Fox, P.L.; Kim, S. Aminoacyl-TRNA Synthetases as Therapeutic Targets. Nat. Rev. Drug Discov. 2019, 18, 629–650. [Google Scholar] [CrossRef]
- Kim, S.-H.; Bae, S.; Song, M. Recent Development of Aminoacyl-TRNA Synthetase Inhibitors for Human Diseases: A Future Perspective. Biomolecules 2020, 10, 1625. [Google Scholar] [CrossRef]
- Kanamaru, T.; Nakano, Y.; Toyoda, Y.; Miyagawa, K.-I.; Tada, M.; Kaisho, T.; Nakao, M. In Vitro and In Vivo Antibacterial Activities of TAK-083, an Agent for Treatment of Helicobacter Pylori Infection. Antimicrob. Agents Chemother. 2001, 45, 2455–2459. [Google Scholar] [CrossRef] [PubMed]
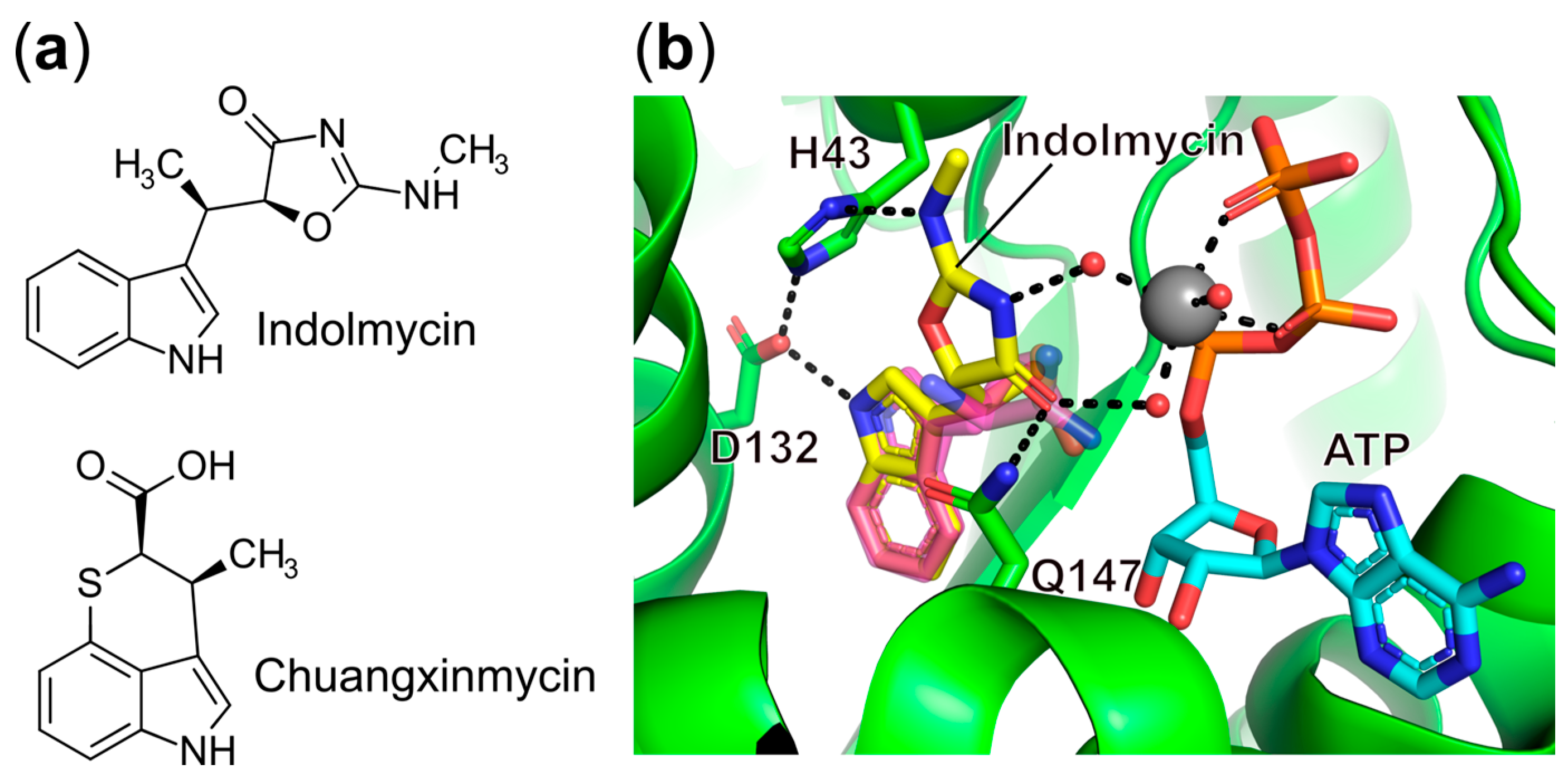
- Williams, T.L.; Yin, Y.W.; Carter, C.W. Selective Inhibition of Bacterial Tryptophanyl-TRNA Synthetases by Indolmycin Is Mechanism-Based. J. Biol. Chem. 2016, 291, 255–265. [Google Scholar] [CrossRef]
- Weinreb, V.; Li, L.; Carter, C.W. A Master Switch Couples Mg2+-Assisted Catalysis to Domain Motion in B. Stearothermophilus Tryptophanyl-TRNA Synthetase. Structure 2012, 20, 128–138. [Google Scholar] [CrossRef]
- Shen, N.; Zhou, M.; Yang, B.; Yu, Y.; Dong, X.; Ding, J. Catalytic Mechanism of the Tryptophan Activation Reaction Revealed by Crystal Structures of Human Tryptophanyl-TRNA Synthetase in Different Enzymatic States. Nucleic Acids Res. 2008, 36, 1288–1299. [Google Scholar] [CrossRef]
- Werner, R.G.; Reuter, W. Interaction of Indolmycin in the Metabolism of Tryptophan in Rat Liver. Arzneimittelforschung 1979, 29, 59–63. [Google Scholar]
- Brown, M.J.; Carter, P.S.; Fenwick, A.S.; Fosberry, A.P.; Hamprecht, D.W.; Hibbs, M.J.; Jarvest, R.L.; Mensah, L.; Milner, P.H.; O’Hanlon, P.J.; et al. The Antimicrobial Natural Product Chuangxinmycin and Some Synthetic Analogues Are Potent and Selective Inhibitors of Bacterial Tryptophanyl TRNA Synthetase. Bioorg. Med. Chem. Lett. 2002, 12, 3171–3174. [Google Scholar] [CrossRef]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for Aminoacyl-TRNA Synthetase Inhibitors as New Antimicrobial Agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef]
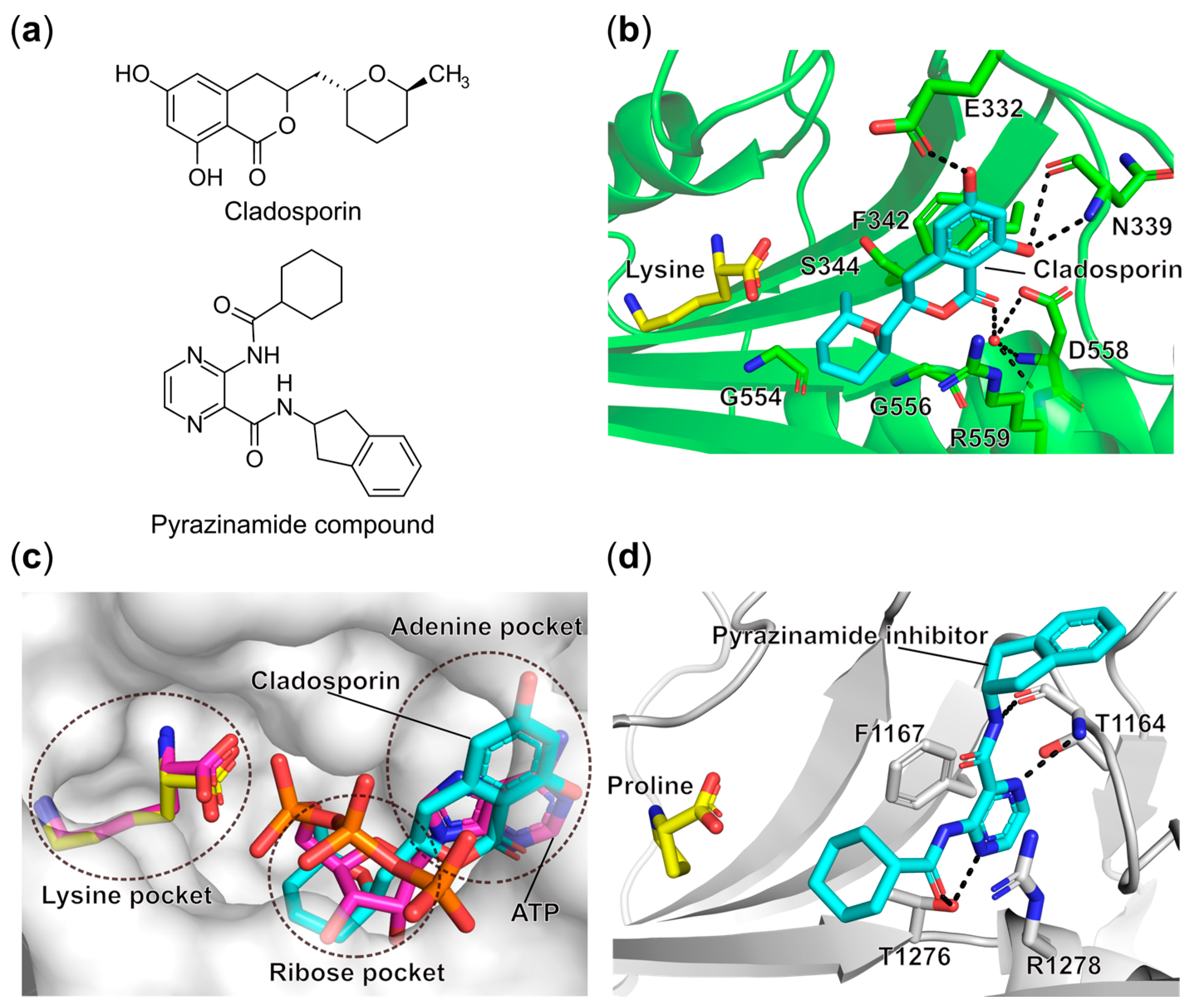
- Scott, P.M.; Van Walbeek, W.; MacLean, W.M. Cladosporin, a New Antifungal Metabolite from Cladosporium Cladosporioides. J. Antibiot. 1971, 24, 747–755. [Google Scholar] [CrossRef]
- Hoepfner, D.; McNamara, C.W.; Lim, C.S.; Studer, C.; Riedl, R.; Aust, T.; McCormack, S.L.; Plouffe, D.M.; Meister, S.; Schuierer, S.; et al. Selective and Specific Inhibition of the Plasmodium Falciparum Lysyl-TRNA Synthetase by the Fungal Secondary Metabolite Cladosporin. Cell Host Microbe 2012, 11, 654–663. [Google Scholar] [CrossRef]
- Khan, S.; Sharma, A.; Belrhali, H.; Yogavel, M.; Sharma, A. Structural Basis of Malaria Parasite Lysyl-TRNA Synthetase Inhibition by Cladosporin. J. Struct. Funct. Genom. 2014, 15, 63–71. [Google Scholar] [CrossRef]
- Fang, P.; Han, H.; Wang, J.; Chen, K.; Chen, X.; Guo, M. Structural Basis for Specific Inhibition of TRNA Synthetase by an ATP Competitive Inhibitor. Chem. Biol. 2015, 22, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Baragaña, B.; Forte, B.; Choi, R.; Hewitt, S.N.; Bueren-Calabuig, J.A.; Pisco, J.P.; Peet, C.; Dranow, D.M.; Robinson, D.A.; Jansen, C.; et al. Lysyl-TRNA Synthetase as a Drug Target in Malaria and Cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2019, 116, 7015–7020. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, L.; Hei, Z.; Li, W.; Wang, J.; Yu, B.; Fang, P. Atomic Resolution Analyses of Isocoumarin Derivatives for Inhibition of Lysyl-TRNA Synthetase. ACS Chem. Biol. 2020, 15, 1016–1025. [Google Scholar] [CrossRef]
- Adachi, R.; Okada, K.; Skene, R.; Ogawa, K.; Miwa, M.; Tsuchinaga, K.; Ohkubo, S.; Henta, T.; Kawamoto, T. Discovery of a Novel Prolyl-TRNA Synthetase Inhibitor and Elucidation of Its Binding Mode to the ATP Site in Complex with l-Proline. Biochem. Biophys. Res. Commun. 2017, 488, 393–399. [Google Scholar] [CrossRef]
- Shibata, A.; Kuno, M.; Adachi, R.; Sato, Y.; Hattori, H.; Matsuda, A.; Okuzono, Y.; Igaki, K.; Tominari, Y.; Takagi, T.; et al. Discovery and Pharmacological Characterization of a New Class of Prolyl-TRNA Synthetase Inhibitor for Anti-Fibrosis Therapy. PLoS ONE 2017, 12, e0186587. [Google Scholar] [CrossRef]
- Koepfli, J.B.; Mead, J.F.; Brockman, J.A. An Alkaloid with High Antimalarial Activity from Dichroa Febrifuga. J. Am. Chem. Soc. 1947, 69, 1837. [Google Scholar] [CrossRef]
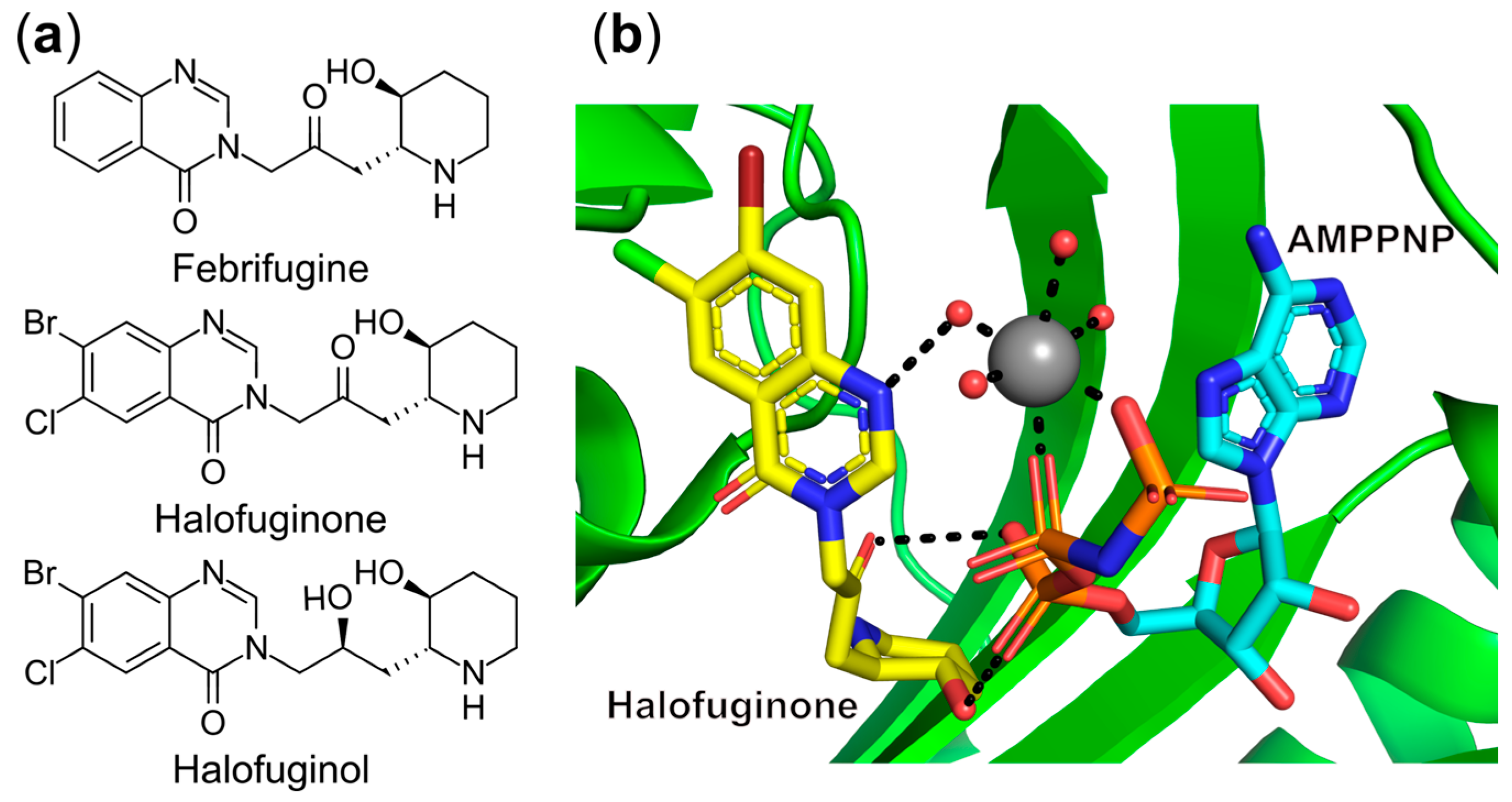
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.-J.; Lee, H.-K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and Other Febrifugine Derivatives Inhibit Prolyl-TRNA Synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef]
- Jain, V.; Yogavel, M.; Oshima, Y.; Kikuchi, H.; Touquet, B.; Hakimi, M.-A.; Sharma, A. Structure of Prolyl-TRNA Synthetase-Halofuginone Complex Provides Basis for Development of Drugs against Malaria and Toxoplasmosis. Structure 2015, 23, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.D.; Pepper, L.R.; Cortese, J.F.; Estiu, G.; Galinsky, K.; Zuzarte-Luis, V.; Derbyshire, E.R.; Ribacke, U.; Lukens, A.K.; Santos, S.A.; et al. The Cytoplasmic Prolyl-TRNA Synthetase of the Malaria Parasite Is a Dual-Stage Target of Febrifugine and Its Analogs. Sci. Transl. Med. 2015, 7, 288ra77. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, Z.; Zheng, L.; Hei, Z.; Wang, Z.; Yu, B.; Jiang, L.; Wang, J.; Fang, P. Inhibition of Plasmodium Falciparum Lysyl-TRNA Synthetase via an Anaplastic Lymphoma Kinase Inhibitor. Nucleic Acids Res. 2020, 48, 11566–11576. [Google Scholar] [CrossRef]
- Gentry, D.R.; Ingraham, K.A.; Stanhope, M.J.; Rittenhouse, S.; Jarvest, R.L.; O’Hanlon, P.J.; Brown, J.R.; Holmes, D.J. Variable Sensitivity to Bacterial Methionyl-TRNA Synthetase Inhibitors Reveals Subpopulations of Streptococcus Pneumoniae with Two Distinct Methionyl-TRNA Synthetase Genes. Antimicrob. Agents Chemother. 2003, 47, 1784–1789. [Google Scholar] [CrossRef]
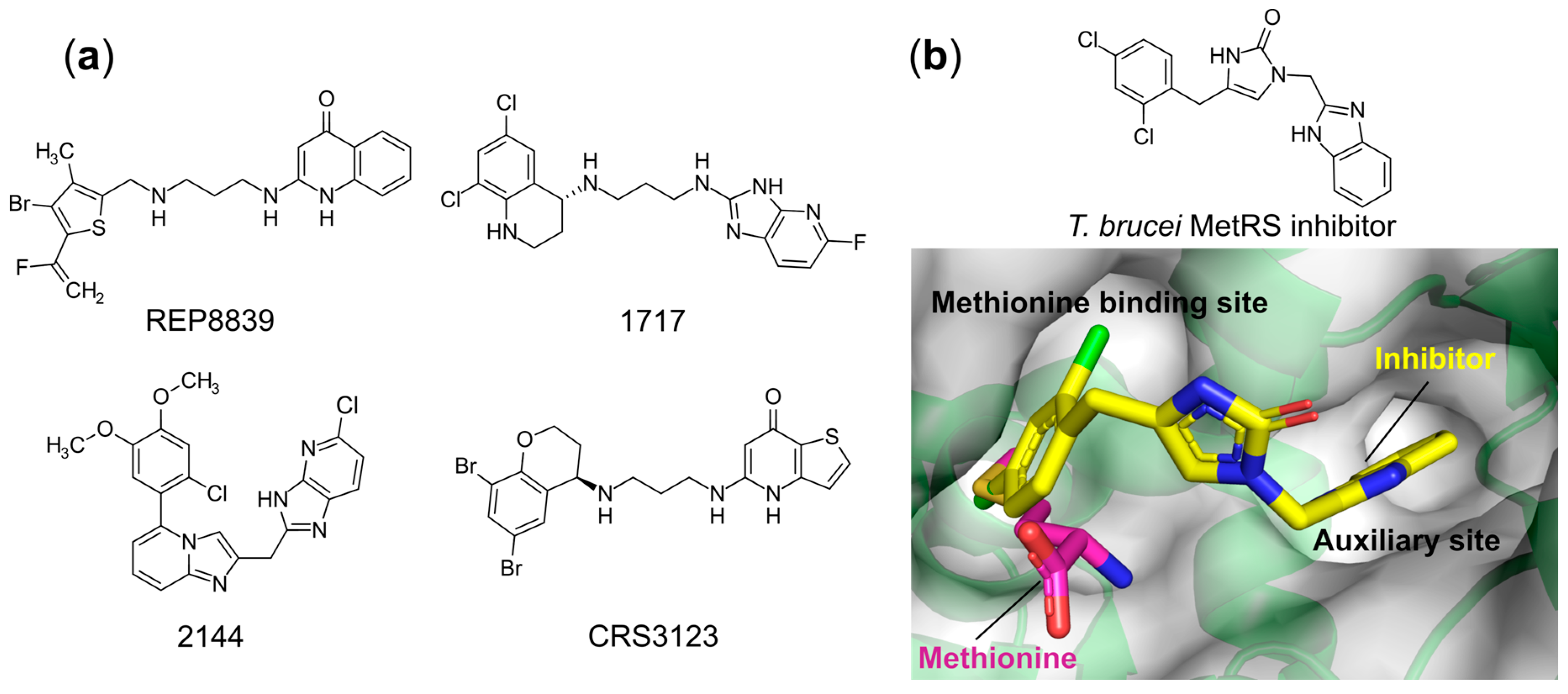
- Critchley, I.A.; Ochsner, U.A. Recent Advances in the Preclinical Evaluation of the Topical Antibacterial Agent REP8839. Curr. Opin. Chem. Biol. 2008, 12, 409–417. [Google Scholar] [CrossRef]
- Ochsner, U.A.; Young, C.L.; Stone, K.C.; Dean, F.B.; Janjic, N.; Critchley, I.A. Mode of Action and Biochemical Characterization of REP8839, a Novel Inhibitor of Methionyl-TRNA Synthetase. Antimicrob. Agents Chemother. 2005, 49, 4253–4262. [Google Scholar] [CrossRef]
- Green, L.S.; Bullard, J.M.; Ribble, W.; Dean, F.; Ayers, D.F.; Ochsner, U.A.; Janjic, N.; Jarvis, T.C. Inhibition of Methionyl-TRNA Synthetase by REP8839 and Effects of Resistance Mutations on Enzyme Activity. Antimicrob. Agents Chemother. 2009, 53, 86–94. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Faghih, O.; Zhang, Z.; Ranade, R.M.; Gillespie, J.R.; Creason, S.A.; Huang, W.; Shibata, S.; Barros-Álvarez, X.; Verlinde, C.L.M.J.; Hol, W.G.J.; et al. Development of Methionyl-TRNA Synthetase Inhibitors as Antibiotics for Gram-Positive Bacterial Infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Nayak, S.U.; Griffiss, J.M.; Blumer, J.; O’Riordan, M.A.; Gray, W.; McKenzie, R.; Jurao, R.A.; An, A.T.; Le, M.; Bell, S.J.; et al. Safety, Tolerability, Systemic Exposure, and Metabolism of CRS3123, a Methionyl-TRNA Synthetase Inhibitor Developed for Treatment of Clostridium Difficile, in a Phase 1 Study. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Lomeli, B.K.; Galbraith, H.; Schettler, J.; Saviolakis, G.A.; El-Amin, W.; Osborn, B.; Ravel, J.; Hazleton, K.; Lozupone, C.A.; Evans, R.J.; et al. Multiple-Ascending-Dose Phase 1 Clinical Study of the Safety, Tolerability, and Pharmacokinetics of CRS3123, a Narrow-Spectrum Agent with Minimal Disruption of Normal Gut Microbiota. Antimicrob. Agents Chemother. 2019, 64. [Google Scholar] [CrossRef]
- Shibata, S.; Gillespie, J.R.; Kelley, A.M.; Napuli, A.J.; Zhang, Z.; Kovzun, K.V.; Pefley, R.M.; Lam, J.; Zucker, F.H.; Van Voorhis, W.C.; et al. Selective Inhibitors of Methionyl-TRNA Synthetase Have Potent Activity against Trypanosoma Brucei Infection in Mice. Antimicrob. Agents Chemother. 2011, 55, 1982–1989. [Google Scholar] [CrossRef]
- Torrie, L.S.; Brand, S.; Robinson, D.A.; Ko, E.J.; Stojanovski, L.; Simeons, F.R.C.; Wyllie, S.; Thomas, J.; Ellis, L.; Osuna-Cabello, M.; et al. Chemical Validation of Methionyl-TRNA Synthetase as a Druggable Target in Leishmania Donovani. ACS Infect. Dis. 2017, 3, 718–727. [Google Scholar] [CrossRef]
- Devine, W.G.; Diaz-Gonzalez, R.; Ceballos-Perez, G.; Rojas, D.; Satoh, T.; Tear, W.; Ranade, R.M.; Barros-Álvarez, X.; Hol, W.G.J.; Buckner, F.S.; et al. From Cells to Mice to Target: Characterization of NEU-1053 (SB-443342) and Its Analogues for Treatment of Human African Trypanosomiasis. ACS Infect. Dis. 2017, 3, 225–236. [Google Scholar] [CrossRef]
- Zhang, Z.; Koh, C.Y.; Ranade, R.M.; Shibata, S.; Gillespie, J.R.; Hulverson, M.A.; Huang, W.; Nguyen, J.; Pendem, N.; Gelb, M.H.; et al. 5-Fluoroimidazo[4,5-b]Pyridine Is a Privileged Fragment That Conveys Bioavailability to Potent Trypanosomal Methionyl-TRNA Synthetase Inhibitors. ACS Infect. Dis. 2016, 2, 399–404. [Google Scholar] [CrossRef]
- Zhang, Z.; Barros-Álvarez, X.; Gillespie, J.R.; Ranade, R.M.; Huang, W.; Shibata, S.; Molasky, N.M.R.; Faghih, O.; Mushtaq, A.; Choy, R.K.M.; et al. Structure-Guided Discovery of Selective Methionyl-TRNA Synthetase Inhibitors with Potent Activity against Trypanosoma Brucei. RSC Med. Chem. 2020, 11, 885–895. [Google Scholar] [CrossRef]
- Koh, C.Y.; Kim, J.E.; Shibata, S.; Ranade, R.M.; Yu, M.; Liu, J.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.M.J.; Fan, E.; et al. Distinct States of Methionyl-TRNA Synthetase Indicate Inhibitor Binding by Conformational Selection. Structure 2012, 20, 1681–1691. [Google Scholar] [CrossRef]
- Fuller, A.T.; Mellows, G.; Woolford, M.; Banks, G.T.; Barrow, K.D.; Chain, E.B. Pseudomonic Acid: An Antibiotic Produced by Pseudomonas Fluorescens. Nature 1971, 234, 416–417. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Lee, J.T.; Wu, H.C.; Kawakami, M. Relationship of Protein Structure of Isoleucyl-TRNA Synthetase with Pseudomonic Acid Resistance of Escherichia Coli. A Proposed Mode of Action of Pseudomonic Acid as an Inhibitor of Isoleucyl-TRNA Synthetase. J. Biol. Chem. 1994, 269, 24304–24309. [Google Scholar] [CrossRef]
- Sutherland, R.; Boon, R.J.; Griffin, K.E.; Masters, P.J.; Slocombe, B.; White, A.R. Antibacterial Activity of Mupirocin (Pseudomonic Acid), a New Antibiotic for Topical Use. Antimicrob. Agents Chemother. 1985, 27, 495–498. [Google Scholar] [CrossRef]
- Hughes, J.; Mellows, G. On the Mode of Action of Pseudomonic Acid: Inhibition of Protein Synthesis in Staphylococcus Aureus. J. Antibiot. 1978, 31, 330–335. [Google Scholar] [CrossRef]
- Hughes, J.; Mellows, G. Interaction of Pseudomonic Acid A with Escherichia Coli B Isoleucyl-TRNA Synthetase. Biochem. J. 1980, 191, 209–219. [Google Scholar] [CrossRef]
- Antonio, M.; McFerran, N.; Pallen, M.J. Mutations Affecting the Rossman Fold of Isoleucyl-TRNA Synthetase Are Correlated with Low-Level Mupirocin Resistance in Staphylococcus Aureus. Antimicrob. Agents Chemother. 2002, 46, 438–442. [Google Scholar] [CrossRef]
- Gilbart, J.; Perry, C.R.; Slocombe, B. High-Level Mupirocin Resistance in Staphylococcus Aureus: Evidence for Two Distinct Isoleucyl-TRNA Synthetases. Antimicrob. Agents Chemother. 1993, 37, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, H.; Kagasaki, T.; Kinoshita, T.; Haruyama, H.; Domon, H.; Utsui, Y.; Kodama, K.; Takahashi, S. Thiomarinol, a New Hybrid Antimicrobial Antibiotic Produced by a Marine Bacterium. Fermentation, Isolation, Structure, and Antimicrobial Activity. J. Antibiot. 1993, 46, 1834–1842. [Google Scholar] [CrossRef]
- Shiozawa, H.; Kagasaki, T.; Torikata, A.; Tanaka, N.; Fujimoto, K.; Hata, T.; Furukawa, Y.; Takahashi, S. Thiomarinols B and C, New Antimicrobial Antibiotics Produced by a Marine Bacterium. J. Antibiot. 1995, 48, 907–909. [Google Scholar] [CrossRef]
- Gamo, F.-J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, J.-L.; Vanderwall, D.E.; Green, D.V.S.; Kumar, V.; Hasan, S.; et al. Thousands of Chemical Starting Points for Antimalarial Lead Identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Mensah, L.M.; Doyle, M.L.; Broom, N.J.; Osbourne, N.; Forrest, A.K.; Richardson, C.M.; O’Hanlon, P.J.; Pope, A.J. Rational Design of Femtomolar Inhibitors of Isoleucyl TRNA Synthetase from a Binding Model for Pseudomonic Acid-A. Biochemistry 2000, 39, 6003–6011. [Google Scholar] [CrossRef]
- Gause, G.F. Recent Studies on Albomycin, a New Antibiotic. Br. Med. J. 1955, 2, 1177–1179. [Google Scholar] [CrossRef]
- Pramanik, A.; Stroeher, U.; Krejci, J.; Standish, A.; Bohn, E.; Paton, J.; Autenrieth, I.; Braun, V. Albomycin Is an Effective Antibiotic, as Exemplified with Yersinia Enterocolitica and Streptococcus Pneumoniae. Int. J. Med. Microbiol. 2007, 297, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, A.L.; Fulston, M.; Houge-Frydrych, C.S.; Jones, J.J.; Warr, S.R. A Potent Seryl TRNA Synthetase Inhibitor SB-217452 Isolated from a Streptomyces Species. J. Antibiot. 2000, 53, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Kulkarni, A.; Yang, Z.; Patil, P.B.; Zhou, W.; Chi, X.; Van Lanen, S.; Chen, S. Biosynthesis of Albomycin Δ2 Provides a Template for Assembling Siderophore and Aminoacyl-TRNA Synthetase Inhibitor Conjugates. ACS Chem. Biol. 2012, 7, 1565–1575. [Google Scholar] [CrossRef]
- Lin, Z.; Xu, X.; Zhao, S.; Yang, X.; Guo, J.; Zhang, Q.; Jing, C.; Chen, S.; He, Y. Total Synthesis and Antimicrobial Evaluation of Natural Albomycins against Clinical Pathogens. Nat. Commun. 2018, 9, 3445. [Google Scholar] [CrossRef]
- Gadakh, B.; Vondenhoff, G.; Pang, L.; Nautiyal, M.; De Graef, S.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Synthesis and Structural Insights into the Binding Mode of the Albomycin Δ1 Core and Its Analogues in Complex with Their Target Aminoacyl-TRNA Synthetase. Bioorg. Med. Chem. 2020, 28, 115645. [Google Scholar] [CrossRef]
- Asensio, C.; Pérez-Díaz, J.C. A New Family of Low Molecular Weight Antibiotics from Enterobacteria. Biochem. Biophys. Res. Commun. 1976, 69, 7–14. [Google Scholar] [CrossRef]
- Baquero, F.; Bouanchaud, D.; Martinez-Perez, M.C.; Fernandez, C. Microcin Plasmids: A Group of Extrachromosomal Elements Coding for Low-Molecular-Weight Antibiotics in Escherichia Coli. J. Bacteriol. 1978, 135, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Metlitskaya, A.; Datsenko, K.; Kazakov, T.; Kazakov, A.; Wanner, B.; Severinov, K. The Escherichia Coli Yej Transporter Is Required for the Uptake of Translation Inhibitor Microcin C. J. Bacteriol. 2007, 189, 8361–8365. [Google Scholar] [CrossRef] [PubMed]
- Metlitskaya, A.; Kazakov, T.; Kommer, A.; Pavlova, O.; Praetorius-Ibba, M.; Ibba, M.; Krasheninnikov, I.; Kolb, V.; Khmel, I.; Severinov, K. Aspartyl-TRNA Synthetase Is the Target of Peptide Nucleotide Antibiotic Microcin C. J. Biol. Chem. 2006, 281, 18033–18042. [Google Scholar] [CrossRef] [PubMed]
- Van de Vijver, P.; Vondenhoff, G.H.M.; Kazakov, T.S.; Semenova, E.; Kuznedelov, K.; Metlitskaya, A.; Van Aerschot, A.; Severinov, K. Synthetic Microcin C Analogs Targeting Different Aminoacyl-TRNA Synthetases. J. Bacteriol. 2009, 191, 6273–6280. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.M.; Dubiley, S.; Severinov, K.; Lescrinier, E.; Rozenski, J.; Van Aerschot, A. Extended Targeting Potential and Improved Synthesis of Microcin C Analogs as Antibacterials. Bioorg. Med. Chem. 2011, 19, 5462–5467. [Google Scholar] [CrossRef]
- Severinov, K.; Nair, S.K. Microcin C: Biosynthesis and Mechanisms of Bacterial Resistance. Future Microbiol. 2012, 7, 281–289. [Google Scholar] [CrossRef]
- Serebryakova, M.; Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Nautiyal, M.; Van Aerschot, A.; Severinov, K.; Dubiley, S. A Trojan-Horse Peptide-Carboxymethyl-Cytidine Antibiotic from Bacillus Amyloliquefaciens. J. Am. Chem. Soc. 2016, 138, 15690–15698. [Google Scholar] [CrossRef]
- Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Piskunova, J.; Severinov, K.; Serebryakova, M.; Dubiley, S. The Product of Yersinia Pseudotuberculosis Mcc Operon Is a Peptide-Cytidine Antibiotic Activated Inside Producing Cells by the TldD/E Protease. J. Am. Chem. Soc. 2017, 139, 16178–16187. [Google Scholar] [CrossRef]
- Kerr, A.; Htay, K. Biological Control of Crown Gall through Bacteriocin Production. Physiol. Plant. Pathol. 1974, 4, 37–44. [Google Scholar] [CrossRef]
- Kerr, A.; Tate, M.E. Agrocins and the Biological Control of Crown Gall. Microbiol. Sci. 1984, 1, 1–4. [Google Scholar] [PubMed]
- Murphy, P.J.; Tate, M.E.; Kerr, A. Substituents at N6 and C-5′ Control Selective Uptake and Toxicity of the Adenine-Nucleotide Bacteriocin, Agrocin 84, in Agrobacteria. Eur. J. Biochem. 1981, 115, 539–543. [Google Scholar] [CrossRef]
- Ryder, M.H.; Tate, M.E.; Jones, G.P. Agrocinopine A, a Tumor-Inducing Plasmid-Coded Enzyme Product, Is a Phosphodiester of Sucrose and L-Arabinose. J. Biol. Chem. 1984, 259, 9704–9710. [Google Scholar] [CrossRef]
- Chopra, S.; Palencia, A.; Virus, C.; Tripathy, A.; Temple, B.R.; Velazquez-Campoy, A.; Cusack, S.; Reader, J.S. Plant Tumour Biocontrol Agent Employs a TRNA-Dependent Mechanism to Inhibit Leucyl-TRNA Synthetase. Nat. Commun. 2013, 4, 1417. [Google Scholar] [CrossRef]
- Zeng, Y.; Roy, H.; Patil, P.B.; Ibba, M.; Chen, S. Characterization of Two Seryl-TRNA Synthetases in Albomycin-Producing Streptomyces Sp. Strain ATCC 700974. Antimicrob. Agents Chemother. 2009, 53, 4619–4627. [Google Scholar] [CrossRef]
- Olano, C.; Wilkinson, B.; Sánchez, C.; Moss, S.J.; Sheridan, R.; Math, V.; Weston, A.J.; Braña, A.F.; Martin, C.J.; Oliynyk, M.; et al. Biosynthesis of the Angiogenesis Inhibitor Borrelidin by Streptomyces Parvulus Tü4055: Cluster Analysis and Assignment of Functions. Chem. Biol. 2004, 11, 87–97. [Google Scholar] [CrossRef][Green Version]
- Yanagisawa, T.; Kawakami, M. How Does Pseudomonas Fluorescens Avoid Suicide from Its Antibiotic Pseudomonic Acid? Evidence for Two Evolutionarily Distinct Isoleucyl-TRNA Synthetases Conferring Self-Defense. J. Biol. Chem. 2003, 278, 25887–25894. [Google Scholar] [CrossRef]
- Kitabatake, M.; Ali, K.; Demain, A.; Sakamoto, K.; Yokoyama, S.; Söll, D. Indolmycin Resistance of Streptomyces Coelicolor A3(2) by Induced Expression of One of Its Two Tryptophanyl-TRNA Synthetases. J. Biol. Chem. 2002, 277, 23882–23887. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Palencia, A.; Virus, C.; Schulwitz, S.; Temple, B.R.; Cusack, S.; Reader, J. Structural Characterization of Antibiotic Self-Immunity TRNA Synthetase in Plant Tumour Biocontrol Agent. Nat. Commun. 2016, 7, 12928. [Google Scholar] [CrossRef] [PubMed]
- Bernier, S.; Akochy, P.-M.; Lapointe, J.; Chênevert, R. Synthesis and Aminoacyl-TRNA Synthetase Inhibitory Activity of Aspartyl Adenylate Analogs. Bioorg. Med. Chem. 2005, 13, 69–75. [Google Scholar] [CrossRef]
- Hill, J.M.; Yu, G.; Shue, Y.-K.; Zydowsky, T.M.; Rebek, J. Aminoacyl Adenylate Mimics as Novel Antimicrobial and Antiparasitic Agents. U.S. Patent 5,726,195, 10 March 1998. [Google Scholar]
- De Ruysscher, D.; Pang, L.; Lenders, S.M.G.; Cappoen, D.; Cos, P.; Rozenski, J.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Synthesis and Structure-Activity Studies of Novel Anhydrohexitol-Based Leucyl-TRNA Synthetase Inhibitors. Eur. J. Med. Chem. 2020, 113021. [Google Scholar] [CrossRef]
- Teng, M.; Hilgers, M.T.; Cunningham, M.L.; Borchardt, A.; Locke, J.B.; Abraham, S.; Haley, G.; Kwan, B.P.; Hall, C.; Hough, G.W.; et al. Identification of Bacteria-Selective Threonyl-TRNA Synthetase Substrate Inhibitors by Structure-Based Design. J. Med. Chem. 2013, 56, 1748–1760. [Google Scholar] [CrossRef]
- Charlton, M.H.; Aleksis, R.; Saint-Leger, A.; Gupta, A.; Loza, E.; Ribas de Pouplana, L.; Kaula, I.; Gustina, D.; Madre, M.; Lola, D.; et al. N-Leucinyl Benzenesulfonamides as Structurally Simplified Leucyl-TRNA Synthetase Inhibitors. ACS Med. Chem. Lett. 2018, 9, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.; Salimraj, R.; Punekar, A.S.; Bellini, D.; Fishwick, C.W.G.; Czaplewski, L.; Scott, D.J.; Harris, G.; Dowson, C.G.; Lloyd, A.J.; et al. Structure-Guided Enhancement of Selectivity of Chemical Probe Inhibitors Targeting Bacterial Seryl-TRNA Synthetase. J. Med. Chem. 2019, 62, 9703–9717. [Google Scholar] [CrossRef] [PubMed]
- Gadakh, B.; Smaers, S.; Rozenski, J.; Froeyen, M.; Van Aerschot, A. 5′-(N-Aminoacyl)-Sulfonamido-5′-Deoxyadenosine: Attempts for a Stable Alternative for Aminoacyl-Sulfamoyl Adenosines as AaRS Inhibitors. Eur. J. Med. Chem. 2015, 93, 227–236. [Google Scholar] [CrossRef] [PubMed]
- De Ruysscher, D.; Pang, L.; De Graef, S.; Nautiyal, M.; De Borggraeve, W.M.; Rozenski, J.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Acylated Sulfonamide Adenosines as Potent Inhibitors of the Adenylate-Forming Enzyme Superfamily. Eur. J. Med. Chem. 2019, 174, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; De Graef, S.; Nautiyal, M.; Pang, L.; Gadakh, B.; Froeyen, M.; Van Mellaert, L.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Family-Wide Analysis of Aminoacyl-Sulfamoyl-3-Deazaadenosine Analogues as Inhibitors of Aminoacyl-TRNA Synthetases. Eur. J. Med. Chem. 2018, 148, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, M.; De Graef, S.; Pang, L.; Gadakh, B.; Strelkov, S.V.; Weeks, S.D.; Van Aerschot, A. Comparative Analysis of Pyrimidine Substituted Aminoacyl-Sulfamoyl Nucleosides as Potential Inhibitors Targeting Class I Aminoacyl-TRNA Synthetases. Eur. J. Med. Chem. 2019, 173, 154–166. [Google Scholar] [CrossRef]
- Pang, L.; Nautiyal, M.; De Graef, S.; Gadakh, B.; Zorzini, V.; Economou, A.; Strelkov, S.V.; Van Aerschot, A.; Weeks, S.D. Structural Insights into the Binding of Natural Pyrimidine-Based Inhibitors of Class II Aminoacyl-TRNA Synthetases. ACS Chem. Biol. 2020, 15, 407–415. [Google Scholar] [CrossRef]
- Novikova, M.; Kazakov, T.; Vondenhoff, G.H.; Semenova, E.; Rozenski, J.; Metlytskaya, A.; Zukher, I.; Tikhonov, A.; Van Aerschot, A.; Severinov, K. MccE Provides Resistance to Protein Synthesis Inhibitor Microcin C by Acetylating the Processed Form of the Antibiotic. J. Biol. Chem. 2010, 285, 12662–12669. [Google Scholar] [CrossRef]
- Kazakov, T.; Kuznedelov, K.; Semenova, E.; Mukhamedyarov, D.; Datsenko, K.A.; Metlitskaya, A.; Vondenhoff, G.H.; Tikhonov, A.; Agarwal, V.; Nair, S.; et al. The RimL Transacetylase Provides Resistance to Translation Inhibitor Microcin C. J. Bacteriol. 2014, 196, 3377–3385. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.; Pugach, K.; Gadakh, B.; Carlier, L.; Rozenski, J.; Froeyen, M.; Severinov, K.; Van Aerschot, A. N-Alkylated Aminoacyl Sulfamoyladenosines as Potential Inhibitors of Aminoacylation Reactions and Microcin C Analogues Containing D-Amino Acids. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Berger, J.; Jampolsky, L.M.; Goldberg, M.W. Borrelidin, a New Antibiotic with Antiborrelia Activity and Penicillin Enhancement Properties. Arch. Biochem. 1949, 22, 476–478. [Google Scholar] [PubMed]
- Lumb, M.; Macey, P.E.; Spyvee, J.; Whitmarsh, J.M.; Wright, R.D. Isolation of Vivomycin and Borrelidin, Two Antibiotics with Anti-Viral Activity, from a Species of Streptomyces (C 2989). Nature 1965, 206, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-X.; Zhang, J.; Wang, X.-J.; Qian, P.-T.; Wang, J.-D.; Gao, Y.-M.; Yan, Y.-J.; Zhang, S.-Z.; Xu, P.-F.; Li, W.-B.; et al. Antifungal Activity of Borrelidin Produced by a Streptomyces Strain Isolated from Soybean. J. Agric. Food Chem. 2012, 60, 1251–1257. [Google Scholar] [CrossRef]
- Otoguro, K.; Ui, H.; Ishiyama, A.; Kobayashi, M.; Togashi, H.; Takahashi, Y.; Masuma, R.; Tanaka, H.; Tomoda, H.; Yamada, H.; et al. In Vitro and in Vivo Antimalarial Activities of a Non-Glycosidic 18-Membered Macrolide Antibiotic, Borrelidin, against Drug-Resistant Strains of Plasmodia. J. Antibiot. 2003, 56, 727–729. [Google Scholar] [CrossRef]
- Novoa, E.M.; Camacho, N.; Tor, A.; Wilkinson, B.; Moss, S.; Marín-García, P.; Azcárate, I.G.; Bautista, J.M.; Mirando, A.C.; Francklyn, C.S.; et al. Analogs of Natural Aminoacyl-TRNA Synthetase Inhibitors Clear Malaria in Vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E5508–E5517. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, A.; Tanaka, T.; Hirose, T.; Ishiyama, A.; Iwatsuki, M.; Takahashi, Y.; Otoguro, K.; Ōmura, S.; Sunazuka, T. Borrelidin Analogues with Antimalarial Activity: Design, Synthesis and Biological Evaluation against Plasmodium Falciparum Parasites. Bioorg. Med. Chem. Lett. 2013, 23, 2302–2305. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.; Bovee, M.L.; Sacher, M.; Stathopoulos, C.; Poralla, K.; Francklyn, C.S.; Söll, D. A Unique Hydrophobic Cluster near the Active Site Contributes to Differences in Borrelidin Inhibition among Threonyl-TRNA Synthetases. J. Biol. Chem. 2005, 280, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Yu, X.; Jeong, S.J.; Mirando, A.; Chen, K.; Chen, X.; Kim, S.; Francklyn, C.S.; Guo, M. Structural Basis for Full-Spectrum Inhibition of Translational Functions on a TRNA Synthetase. Nat. Commun. 2015, 6, 6402. [Google Scholar] [CrossRef]
- Baker, S.J.; Zhang, Y.-K.; Akama, T.; Lau, A.; Zhou, H.; Hernandez, V.; Mao, W.; Alley, M.R.K.; Sanders, V.; Plattner, J.J. Discovery of a New Boron-Containing Antifungal Agent, 5-Fluoro-1,3-Dihydro-1-Hydroxy-2,1- Benzoxaborole (AN2690), for the Potential Treatment of Onychomycosis. J. Med. Chem. 2006, 49, 4447–4450. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, S. Recent Development of Leucyl-TRNA Synthetase Inhibitors as Antimicrobial Agents. Medchemcomm 2019, 10, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, V.; Crépin, T.; Palencia, A.; Cusack, S.; Akama, T.; Baker, S.J.; Bu, W.; Feng, L.; Freund, Y.R.; Liu, L.; et al. Discovery of a Novel Class of Boron-Based Antibacterials with Activity against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2013, 57, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, K.; Spivak, A.T.; Ingraham, K.; Min, S.; Holmes, D.J.; Jakielaszek, C.; Rittenhouse, S.; Kwan, A.L.; Livi, G.P.; Sathe, G.; et al. Bacterial Resistance to Leucyl-TRNA Synthetase Inhibitor GSK2251052 Develops during Treatment of Complicated Urinary Tract Infections. Antimicrob. Agents Chemother. 2015, 59, 289–298. [Google Scholar] [CrossRef]
- Tenero, D.; Derimanov, G.; Carlton, A.; Tonkyn, J.; Davies, M.; Cozens, S.; Gresham, S.; Gaudion, A.; Puri, A.; Muliaditan, M.; et al. First-Time-in-Human Study and Prediction of Early Bactericidal Activity for GSK3036656, a Potent Leucyl-TRNA Synthetase Inhibitor for Tuberculosis Treatment. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Hao, G.; Li, H.; Yang, F.; Dong, D.; Li, Z.; Ding, Y.; Pan, W.; Wang, E.; Liu, R.; Zhou, H. Discovery of Benzhydrol-Oxaborole Derivatives as Streptococcus Pneumoniae Leucyl-TRNA Synthetase Inhibitors. Bioorg. Med. Chem. 2020, 115871. [Google Scholar] [CrossRef]
- Ho, J.M.; Bakkalbasi, E.; Söll, D.; Miller, C.A. Drugging TRNA Aminoacylation. RNA Biol. 2018, 15, 667–677. [Google Scholar] [CrossRef]
- Mikkelsen, N.E.; Johansson, K.; Virtanen, A.; Kirsebom, L.A. Aminoglycoside Binding Displaces a Divalent Metal Ion in a TRNA-Neomycin B Complex. Nat. Struct. Biol. 2001, 8, 510–514. [Google Scholar] [CrossRef]
- Walter, F.; Pütz, J.; Giegé, R.; Westhof, E. Binding of Tobramycin Leads to Conformational Changes in Yeast TRNA(Asp) and Inhibition of Aminoacylation. EMBO J. 2002, 21, 760–768. [Google Scholar] [CrossRef]
- Wang, W.; Qin, B.; Wojdyla, J.A.; Wang, M.; Gao, X.; Cui, S. Structural Characterization of Free-State and Product-State Mycobacterium Tuberculosis Methionyl-TRNA Synthetase Reveals an Induced-Fit Ligand-Recognition Mechanism. IUCrJ 2018, 5, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.M.; Cai, Y.C.; Spremulli, L.L. Expression and Characterization of the Human Mitochondrial Leucyl-TRNA Synthetase. Biochim. Biophys. Acta 2000, 1490, 245–258. [Google Scholar] [CrossRef]
- Buckner, F.S.; Ranade, R.M.; Gillespie, J.R.; Shibata, S.; Hulverson, M.A.; Zhang, Z.; Huang, W.; Choi, R.; Verlinde, C.L.M.J.; Hol, W.G.J.; et al. Optimization of Methionyl TRNA-Synthetase Inhibitors for Treatment of Cryptosporidium Infection. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Saint-Léger, A.; Ribas de Pouplana, L. A New Set of Assays for the Discovery of Aminoacyl-TRNA Synthetase Inhibitors. Methods 2017, 113, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Benedetto Tiz, D.; Kikelj, D.; Zidar, N. Overcoming Problems of Poor Drug Penetration into Bacteria: Challenges and Strategies for Medicinal Chemists. Expert Opin. Drug Discov. 2018, 13, 497–507. [Google Scholar] [CrossRef]
- Schalk, I.J. A Trojan-Horse Strategy Including a Bacterial Suicide Action for the Efficient Use of a Specific Gram-Positive Antibiotic on Gram-Negative Bacteria. J. Med. Chem. 2018, 61, 3842–3844. [Google Scholar] [CrossRef]
- Dassonville-Klimpt, A.; Sonnet, P. Advances in ‘Trojan Horse’ Strategies in Antibiotic Delivery Systems. Future Med. Chem. 2020, 12, 983–986. [Google Scholar] [CrossRef] [PubMed]
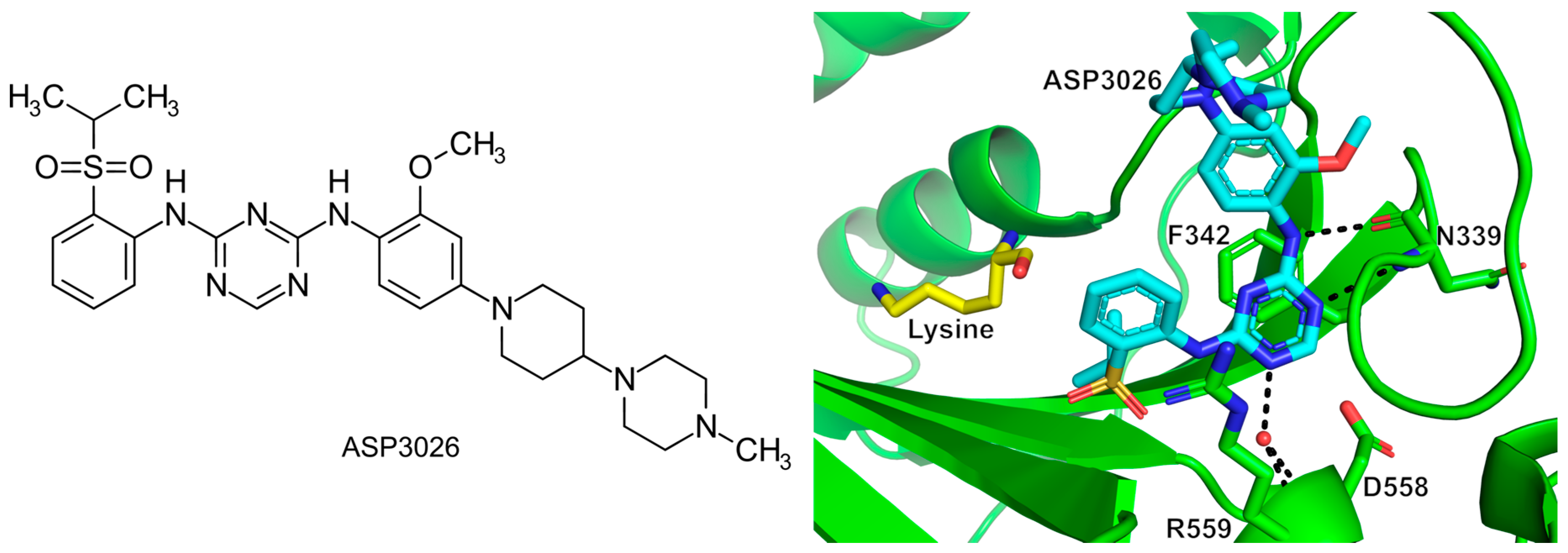
- Randall, C.P.; Rasina, D.; Jirgensons, A.; O’Neill, A.J. Targeting Multiple Aminoacyl-TRNA Synthetases Overcomes the Resistance Liabilities Associated with Antibacterial Inhibitors Acting on a Single Such Enzyme. Antimicrob. Agents Chemother. 2016, 60, 6359–6361. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class I aaRSs | Class II aaRSs | ||||
|---|---|---|---|---|---|
| Ia | Ib | Ic | IIa | IIb | IIc |
| CysRS | ArgRS | TrpRS | GlyRS 2 | AsnRS | AlaRS |
| IleRS | GlnRS | TyrRS | HisRS | AspRS | GlyRS 4 |
| LeuRS | GluRS | ProRS | LysRS 3 | PheRS | |
| MetRS | LysRS 1 | SerRS | PylRS | ||
| ValRS | ThrRS | SepRS | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, L.; Weeks, S.D.; Van Aerschot, A. Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Int. J. Mol. Sci. 2021, 22, 1750. https://doi.org/10.3390/ijms22041750
Pang L, Weeks SD, Van Aerschot A. Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. International Journal of Molecular Sciences. 2021; 22(4):1750. https://doi.org/10.3390/ijms22041750
Chicago/Turabian StylePang, Luping, Stephen D. Weeks, and Arthur Van Aerschot. 2021. "Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery" International Journal of Molecular Sciences 22, no. 4: 1750. https://doi.org/10.3390/ijms22041750
APA StylePang, L., Weeks, S. D., & Van Aerschot, A. (2021). Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. International Journal of Molecular Sciences, 22(4), 1750. https://doi.org/10.3390/ijms22041750