
GEM-Based Metabolic Profiling for Human Bone Osteosarcoma under Different Glucose and Glutamine Availability



Abstract
1. Introduction
- Do nutrient availability changes affect metabolism of the U2OS cells?
- Do changes in glucose and/or glutamine availability impact the growth rate and metabolic requirement of U2OS cells?
- Does simultaneous integration of the gene expression and nutrient availability data into a GEM provide accurate prediction of cell growth and metabolite consumption/production?
- We provide an integrated GEM-based metabolic profiling framework for identification of the nutrient requirements that necessitates using RNA-seq data specific for each condition.
- We prove that genome-scale models like RECON3D provide a great platform for integration of transcript and nutrient availability data in order to create specific models for studying cell metabolism.
- We prove that translation of the relative importance of gene expression and nutrient availability data into the fluxes based on observed experimental feature(s) is a reliable method to build specific models.
- We identified the impact of glucose and glutamine availability on the metabolism and proliferation rate of the U2OS cells.
- We identified that the U2OS cell line is a glutamine-dependent cancer type.
2. Results
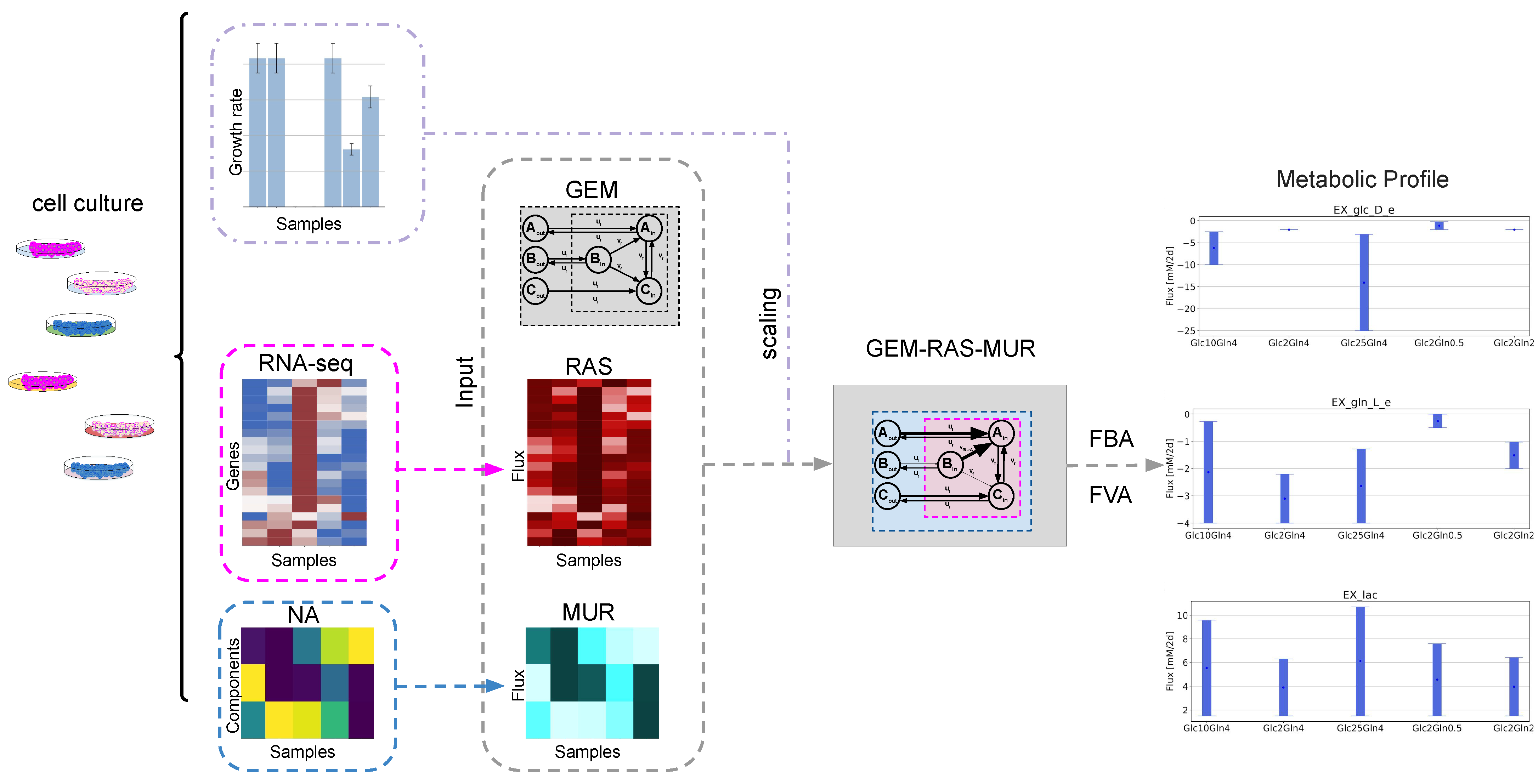
2.1. GEM-Based Metabolism Profiling Methodology
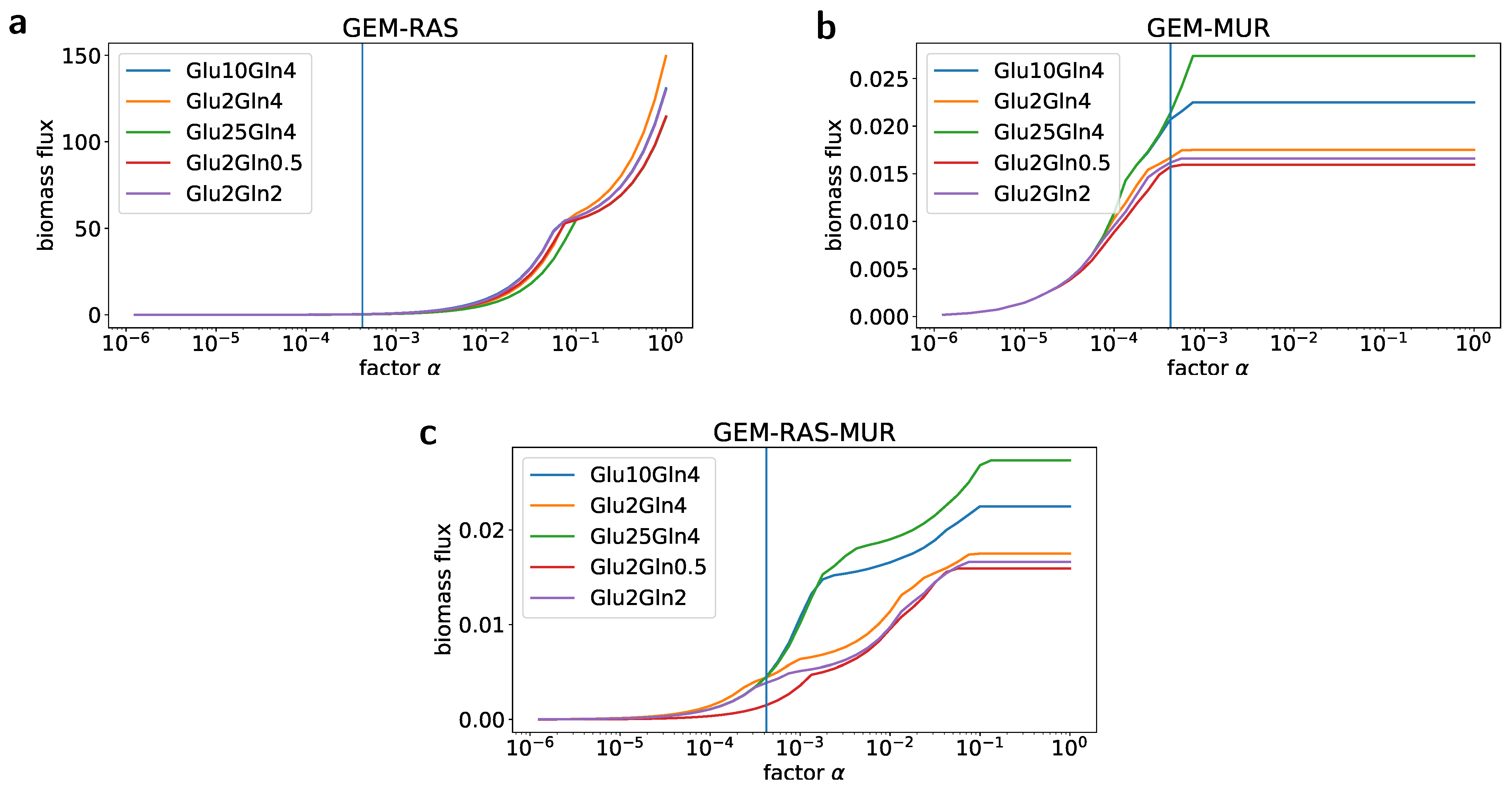
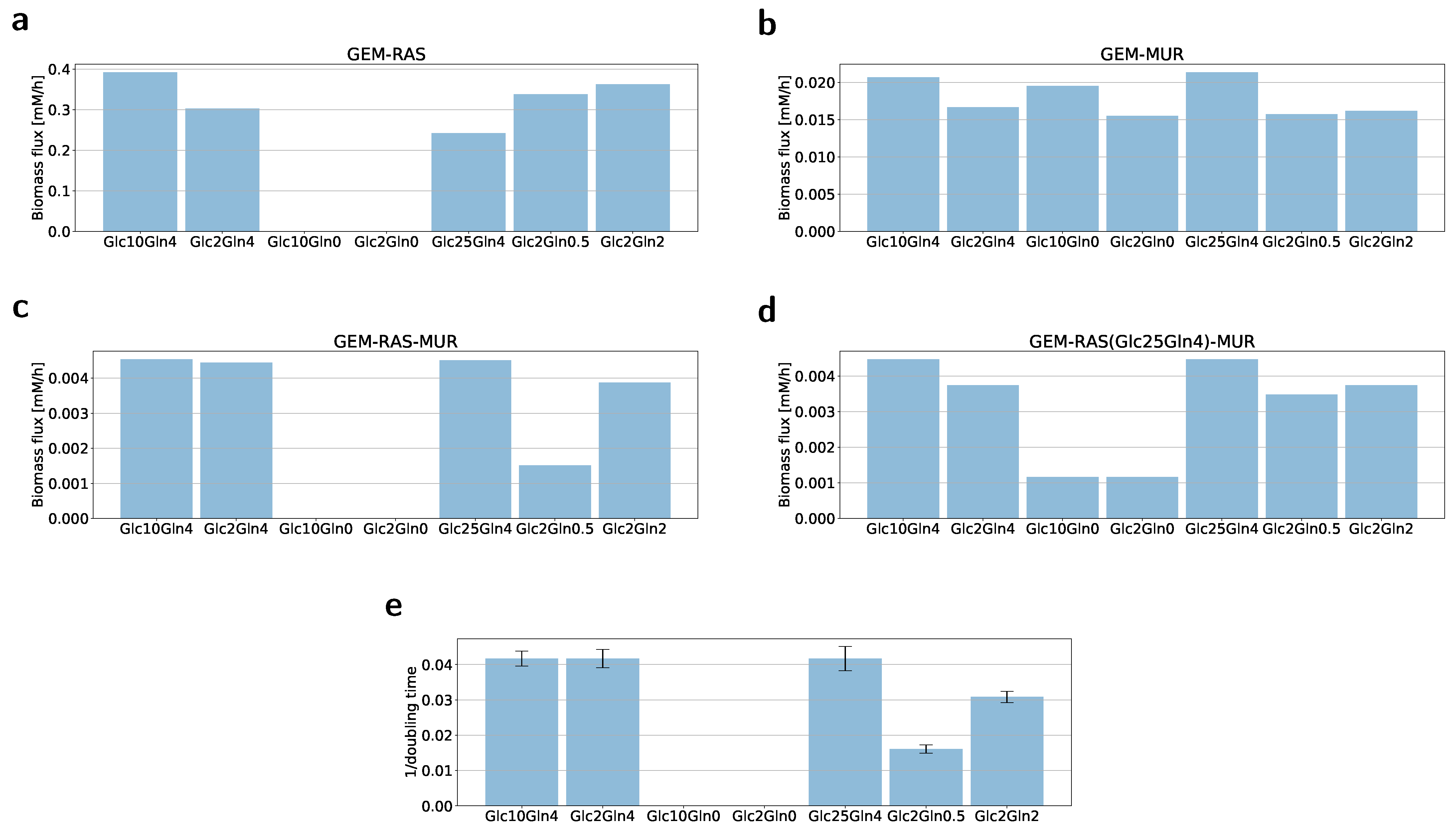
2.2. Formulation of the Specific GEM Model
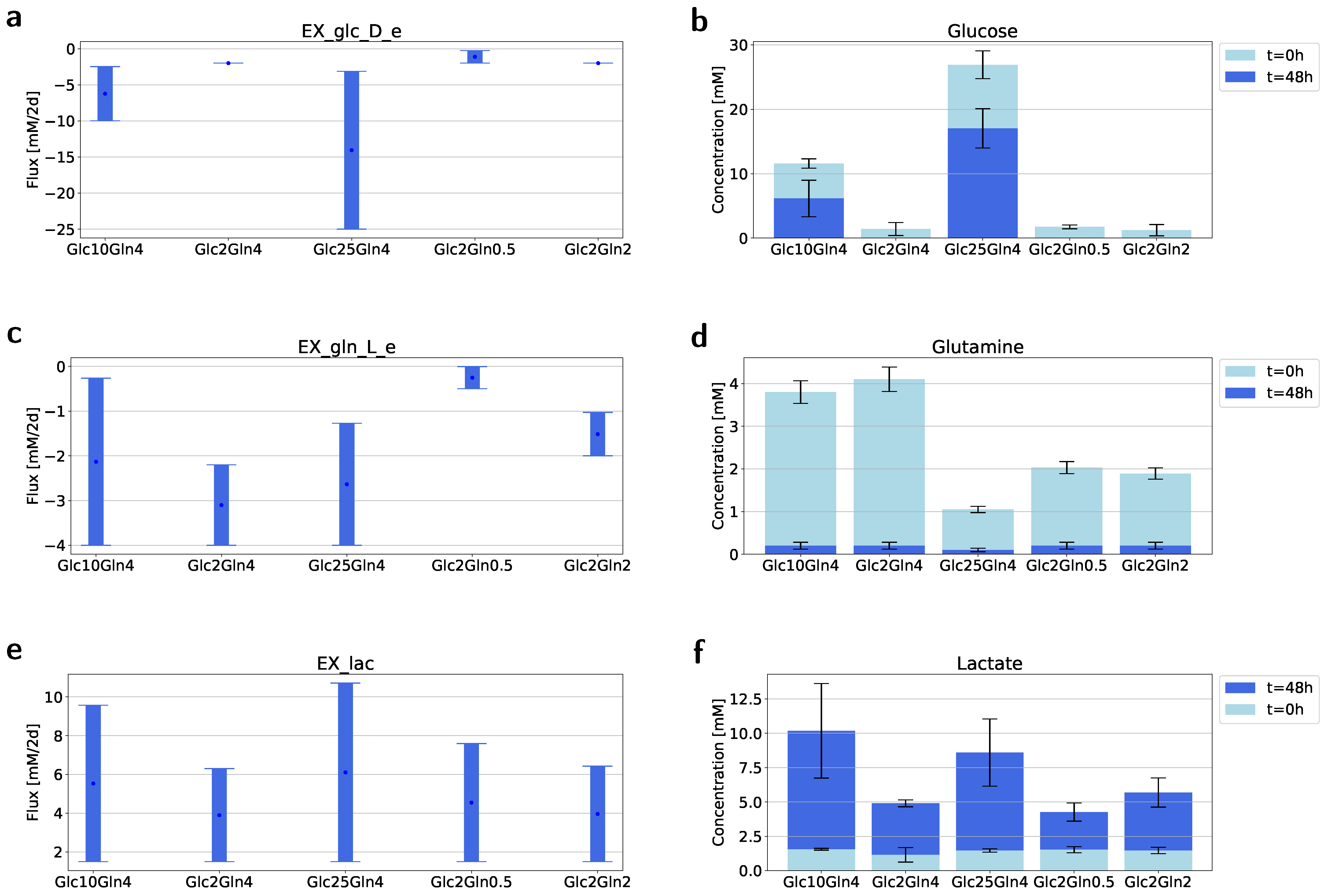
2.3. Flux Variability Analysis to Investigate the Nutrient Requirement
3. Discussion
4. Materials and Methods
4.1. Culturing U2OS Cancer Cells
4.2. Growth Rate/Cell Proliferation Assay
4.3. Isolation and Measurement of the Metabolites from Medium
4.4. Isolation and Obtaining of RNA Data
4.5. RNA-Seq Data Processing
- Sequences were mapped against the human reference genome using Burrows-Wheeler Aligner (BWA; version 0.7.17, https://sourceforge.net/projects/bio-bwa/). The outputs of this step were files in .sam format.
- The .sam files were transformed to sorted bam files using Samtools (https://anaconda.org/bioconda/samtools).
- Gene occurrences were calculated using featuresCounts from the Subread package (https://anaconda.org/bioconda/subread).
- In all cases, the Homo sapiens (human) genome assembly GRCh38 (hg38) from Genome Reference Consortium Build 38 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/) was used as the reference genome.
4.6. Mapping Transcriptomic Data to the Fluxes with Recon3D
4.7. Mapping Nutrient Availability (NA) to Fluxes with Recon3D
4.8. Simulations
4.9. Data and Source Code Availability
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular characterization and clinical relevance of metabolic expression subtypes in human cancers. Cell Rep. 2018, 23, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xia, Y.; Lu, Z. Metabolic features of cancer cells. BMC Cancer Commun. 2018, 38, 65. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Lien, E.C.; Vander Heiden, M.G. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 2019, 19, 651–661. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 2019, 8, e44235. [Google Scholar]
- Muir, A.; Danai, L.V.; Vander Heiden, M.G. Microenvironmental regulation of cancer Cell Metabolism: Implications for experimental design and translational studies. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Gatto, F.; Nielsen, J. Genome-scale modeling of human metabolism–a systems biology approach. Biotechnol. J. 2013, 8, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Prakash, P.; Edwards, J.S. Advances in flux balance analysis. Curr. Opin. Biotechnol. 2003, 14, 491–496. [Google Scholar] [CrossRef]
- Fritsche-Guenther, R.; Gloaguen, Y.; Kirchner, M.; Mertins, P.; Tunn, P.U.; Kirwan, J.A. Progression-dependent altered metabolism in osteosarcoma resulting in different nutrient source dependencies. Cancers 2020, 12, 1371. [Google Scholar] [CrossRef]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Hoang, C.T.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 4. [Google Scholar] [CrossRef]
- Lauvrak, S.U.; Munthe, E.; Kresse, S.H.; Stratford, E.W.; Namløs, H.M.; Meza-Zepeda, L.A.; Myklebost, O. Functional characterisation of osteosarcoma cell lines and identification of mRNAs and miRNAs associated with aggressive cancer phenotypes. Br. J. Cancer 2013, 109, 2228–2236. [Google Scholar] [CrossRef]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Gonzalez, G.A.P.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272. [Google Scholar] [CrossRef] [PubMed]
- Weglarz-Tomczak, E.; Mondeel, T.D.G.A.; Piebes, D.G.E.; Westerhoff, H.V. Simultaneous integration of gene expression and nutrient availability for studying metabolism of hepatocellular carcinoma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Akesson, M.; Forster, J.; Nielsen, J. Integration of gene expression data into genome-scale metabolic models. Metab. Eng. 2004, 6, 285–293. [Google Scholar] [CrossRef]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.-Y.; Moody, D.B.; Murray, M.; Galagan, J.E.; et al. Interpreting expression data with metabolic flux models: Predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef]
- Graudenzi, A.; Maspero, D.; Di Filippo, M.; Gnugnoli, M.; Isella, C.; Mauri, G.; Medico, E.; Antoniotti, M.; Damiani, C. Integration of transcriptomic data and metabolic networks in cancer samples reveals highly significant prognostic power. J. Biomed. Inform. 2018, 87, 37–49. [Google Scholar] [CrossRef]
- Tian, M.; Reed, J.L. Integrating proteomic or transcriptomic data into metabolic models using linear bound flux balance analysis. Bioinformatics 2018, 34, 3882–3888. [Google Scholar] [CrossRef]
- Shlomi, T.; Cabili, M.N.; Herrgard, M.J.; Palsson, B.O.; Ruppin, E. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar] [CrossRef]
- Hyduke, D.R.; Lewis, N.E.; Palsson, B.O. Analysis of omics data with genome-scale models of metabolism. Mol. BioSyst. 2013, 9, 167–174. [Google Scholar] [CrossRef]
- Pandey, V.; Hadadi, N.; Hatzimanikatis, V. Enhanced flux prediction by integrating relative expression and relative metabolite abundance into thermodynamically consistent metabolic models. PLoS Comput. Biol. 2019, 15, e1007036. [Google Scholar] [CrossRef]
- Sapcariu, S.C.; Kanashova, T.; Weindl, D.; Ghelfi, J.; Dittmar, G.; Hiller, K. Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 2014, 1, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models: The COBRA Toolbox v3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition Name | Glucose (mM) | Glutamine (mM) |
|---|---|---|
| Glc10Gln4 | 10 | 4 |
| Glc2Gln4 | 2 | 4 |
| Glc10Gln0 | 10 | 0 |
| Glc2Gln0 | 2 | 0 |
| Glc25Gln4 | 25 | 4 * |
| Glc2Gln0.5 | 2 | 0.5 |
| Glc2Gln2 | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weglarz-Tomczak, E.; Rijlaarsdam, D.J.; Tomczak, J.M.; Brul, S. GEM-Based Metabolic Profiling for Human Bone Osteosarcoma under Different Glucose and Glutamine Availability. Int. J. Mol. Sci. 2021, 22, 1470. https://doi.org/10.3390/ijms22031470
Weglarz-Tomczak E, Rijlaarsdam DJ, Tomczak JM, Brul S. GEM-Based Metabolic Profiling for Human Bone Osteosarcoma under Different Glucose and Glutamine Availability. International Journal of Molecular Sciences. 2021; 22(3):1470. https://doi.org/10.3390/ijms22031470
Chicago/Turabian StyleWeglarz-Tomczak, Ewelina, Demi J. Rijlaarsdam, Jakub M. Tomczak, and Stanley Brul. 2021. "GEM-Based Metabolic Profiling for Human Bone Osteosarcoma under Different Glucose and Glutamine Availability" International Journal of Molecular Sciences 22, no. 3: 1470. https://doi.org/10.3390/ijms22031470
APA StyleWeglarz-Tomczak, E., Rijlaarsdam, D. J., Tomczak, J. M., & Brul, S. (2021). GEM-Based Metabolic Profiling for Human Bone Osteosarcoma under Different Glucose and Glutamine Availability. International Journal of Molecular Sciences, 22(3), 1470. https://doi.org/10.3390/ijms22031470