
HPV Meets APOBEC: New Players in Head and Neck Cancer

 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Head and Neck Squamous Cell Carcinoma
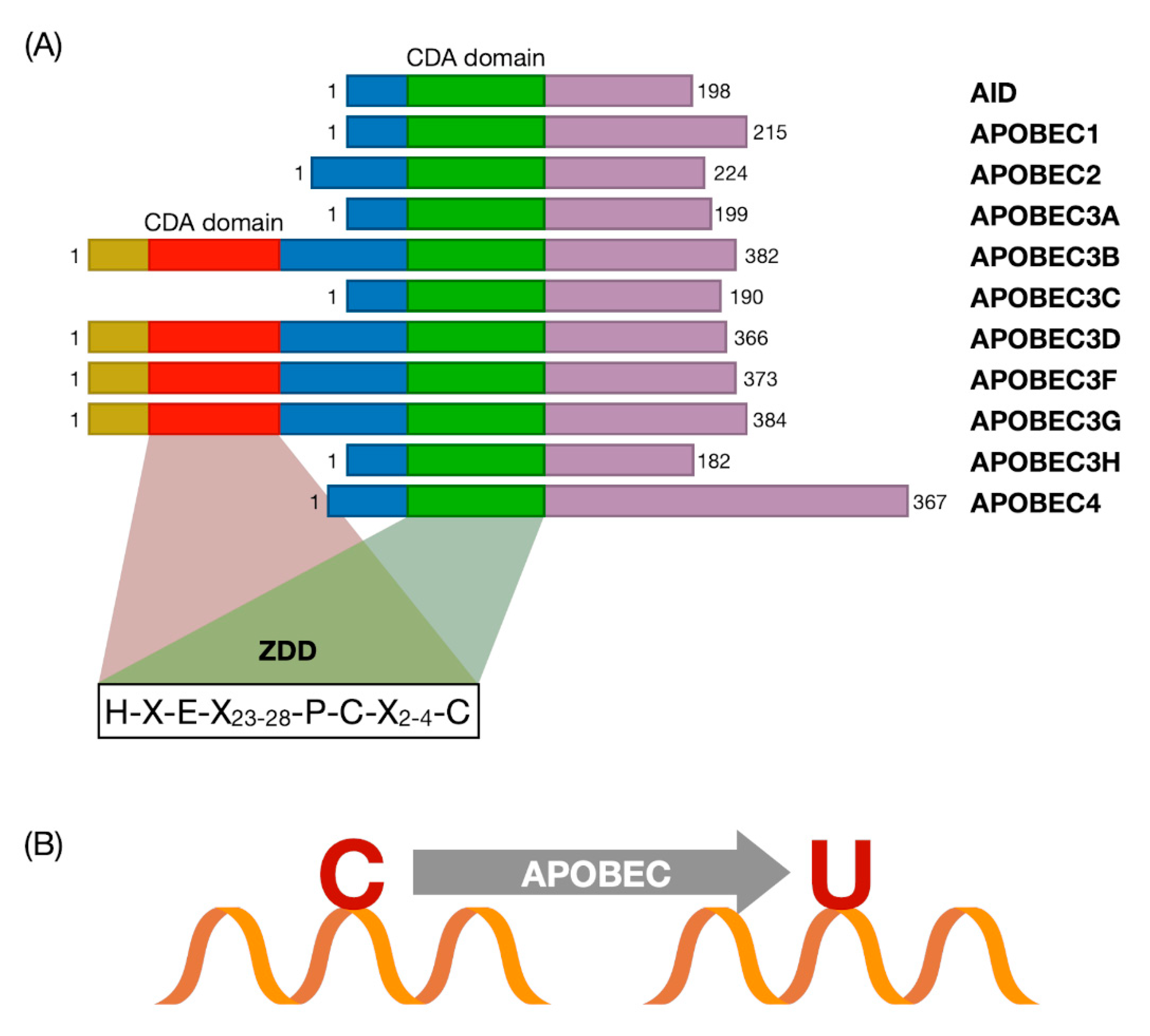
3. The APOBEC Family and Cytosine Deamination
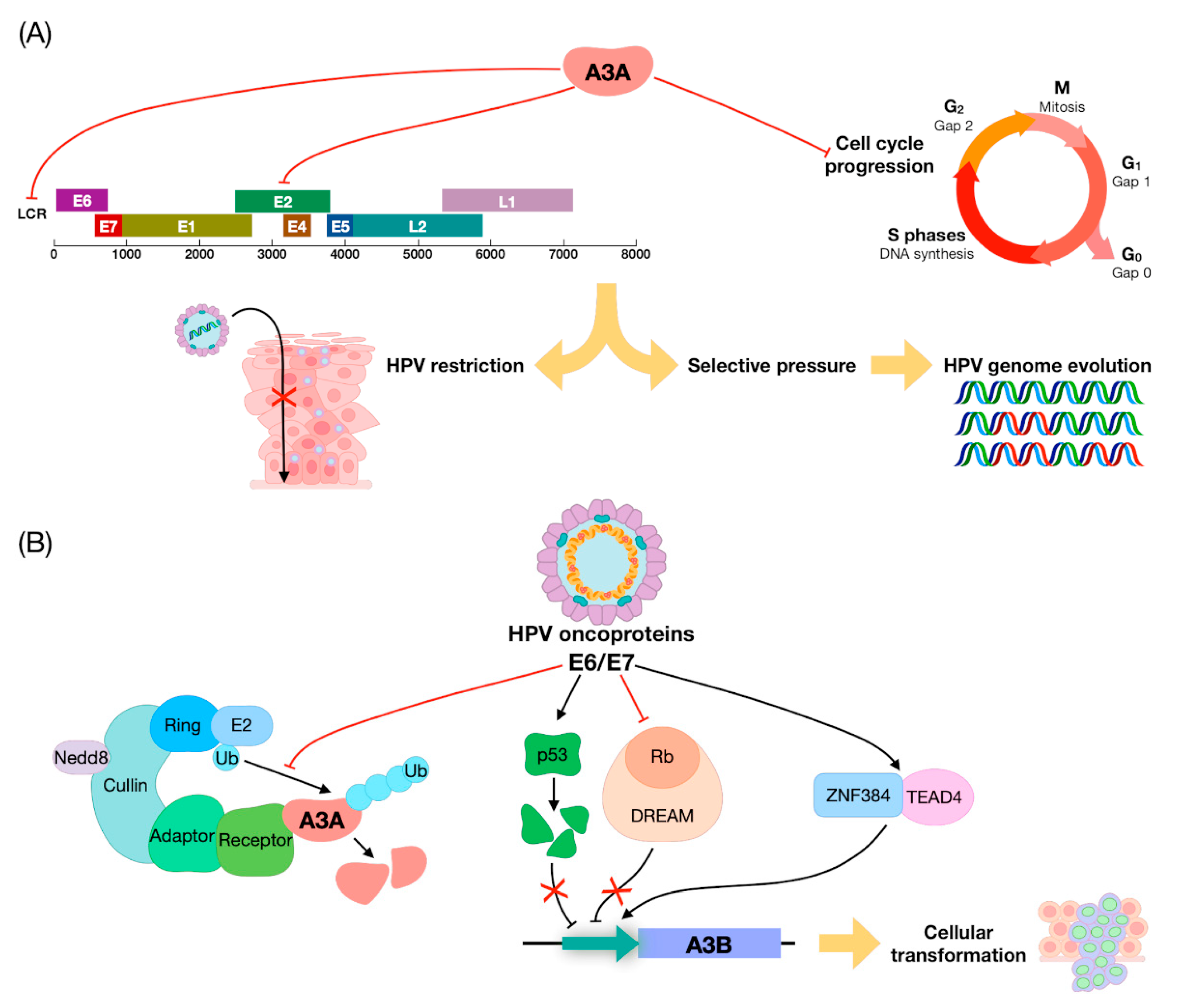
4. APOBEC Genes vs. HPVs: Restriction Factors or DNA Editors?
5. The Role of APOBEC in HNSCC
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McDermott, J.D.; Bowles, D.W. Epidemiology of Head and Neck Squamous Cell Carcinomas: Impact on Staging and Prevention Strategies. Curr. Treat. Options Oncol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Vignot, S.; Lapierre, A.; Falk, A.T.; Guigay, J.; Van Obberghen-Schilling, E.; Milano, G. Integrating genomics in head and neck cancer treatment: Promises and pitfalls. Crit. Rev. Oncol. Hematol. 2015, 95, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Lampri, E.S.; Chondrogiannis, G.; Ioachim, E.; Varouktsi, A.; Mitselou, A.; Galani, A.; Briassoulis, E.; Kanavaros, P.; Galani, V. Biomarkers of head and neck cancer, tools or a gordian knot? Int. J. Clin. Exp. Med. 2015, 8, 10340–10357. [Google Scholar] [PubMed]
- Kuong, K.J.; Loeb, L.A. APOBEC3B mutagenesis in cancer. Nat. Genet. 2013, 45, 964–965. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Faden, D.L.; Thomas, S.; Cantalupo, P.G.; Agrawal, N.; Myers, J.; DeRisi, J. Multi-modality analysis supports APOBEC as a major source of mutations in head and neck squamous cell carcinoma. Oral Oncol. 2017, 74, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 1–10. [Google Scholar] [CrossRef]
- Monjurul, A.M.; Wakae, K.; Wang, Z.; Kitamura, K.; Liu, G.; Koura, M.; Imayasu, M.; Sakamoto, N.; Hanaoka, K.; Nakamura, M.; et al. APOBEC3A and 3C decrease human papillomavirus 16 pseudovirion infectivity. Biochem. Biophys. Res. Commun. 2015, 457, 295–299. [Google Scholar]
- Pautasso, S.; Galitska, G.; Dell’Oste, V.; Biolatti, M.; Cagliani, R.; Forni, D.; De Andrea, M.; Gariglio, M.; Sironi, M.; Landolfo, S. Strategy of Human Cytomegalovirus to Escape Interferon Beta-Induced APOBEC3G Editing Activity. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-Mediated Cytosine Deamination Links PIK3CA Helical Domain Mutations to Human Papillomavirus-Driven Tumor Development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Riva, G.; Pecorari, G.; Biolatti, M.; Pautasso, S.; Lo Cigno, I.; Garzaro, M.; Dell’Oste, V.; Landolfo, S. PYHIN genes as potential biomarkers for prognosis of human papillomavirus-positive or -negative head and neck squamous cell carcinomas. Mol. Biol. Rep. 2019, 46, 3333–3347. [Google Scholar] [CrossRef] [PubMed]
- Riva, G.; Biolatti, M.; Pecorari, G.; Dell’Oste, V.; Landolfo, S. PYHIN Proteins and HPV: Role in the Pathogenesis of Head and Neck Squamous Cell Carcinoma. Microorganisms 2019, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Castellsagué, X.; Alemany, L.; Quer, M.; Halec, G.; Quirós, B.; Tous, S.; Clavero, O.; Alòs, L.; Biegner, T.; Szafarowski, T.; et al. HPV Involvement in Head and Neck Cancers: Comprehensive Assessment of Biomarkers in 3680 Patients. J. Natl. Cancer Inst. 2016, 108, djv403. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systemic review. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef]
- Kang, H.; Kiess, A.; Chung, C.H. Emerging biomarkers in head and neck cancer in the era of genomics. Nat. Rev. Clin. Oncol. 2015, 12, 11–26. [Google Scholar] [CrossRef]
- Braakhuis, B.J.M.; Snijders, P.J.F.; Keune, W.J.H.; Meijer, C.J.L.M.; Ruijter-Schippers, H.J.; Leemans, C.R.; Brakenhoff, R.H. Genetic patterns in head and neck cancers that contain or lack transcriptionally active human papillomavirus. J. Natl. Cancer Inst. 2004, 96, 998–1006. [Google Scholar] [CrossRef]
- Slebos, R.J.C.; Yi, Y.; Ely, K.; Carter, J.; Evjen, A.; Zhang, X.; Shyr, Y.; Murphy, B.M.; Cmelak, A.J.; Burkey, B.B.; et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clin. Cancer Res. 2006, 12, 701–709. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Sahin, A.A.; Gilligan, T.D.; Caudell, J.J. Challenges With the 8th Edition of the AJCC Cancer Staging Manual for Breast, Testicular, and Head and Neck Cancers. J. Natl. Compr. Canc. Netw. 2019, 17, 560–564. [Google Scholar]
- Colevas, A.D.; Yom, S.S.; Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Brizel, D.M.; Burtness, B.; Busse, P.M.; Caudell, J.J.; et al. NCCN guidelines ® insights: Head and neck cancers, version 1.2018 featured updates to the NCCN guidelines. JNCCN J. Natl. Compr. Cancer Netw. 2018, 16, 479–490. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case–Control Study of Human Papillomavirus and Oropharyngeal Cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsagué, X.; Laporte, L.; Bosch, F.X.; de Sanjosé, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Steenbergen, R.D.M.; Snijders, P.J.F.; Heideman, D.A.M.; Meijer, C.J.L.M. Clinical implications of (epi)genetic changes in HPV-induced cervical precancerous lesions. Nat. Rev. Cancer 2014, 14, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Meshman, J.; Wang, P.C.; Chin, R.; John, M.S.; Abemayor, E.; Bhuta, S.; Chen, A.M. Prognostic significance of p16 in squamous cell carcinoma of the larynx and hypopharynx. Am. J. Otolaryngol. Head Neck Med. Surg. 2017, 38, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Urban, D.; Angel, C.; Corry, J.; Lyons, B.; Vallance, N.; Kleid, S.; Iseli, T.A.; Solomon, B.; Rischin, D. Frequency and prognostic significance of p16 INK4A protein overexpression and transcriptionally active human papillomavirus infection in laryngeal squamous cell carcinoma. Br. J. Cancer 2015, 112, 1098–1104. [Google Scholar] [CrossRef]
- Tabor, M.P.; Brakenhoff, R.H.; Ruijter-Schippers, H.J.; Van der Wal, J.E.; Snow, G.B.; Leemans, C.R.; Braakhuis, B.J.M. Multiple head and neck tumors frequently originate from a single preneoplastic lesion. Am. J. Pathol. 2002, 161, 1051–1060. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. EGFR, p16, HPV titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef]
- Alsner, J.; Sørensen, S.B.; Overgaard, J. TP53 mutation is related to poor prognosis after radiotherapy, but not surgery, in squamous cell carcinoma of the head and neck. Radiother. Oncol. 2001, 59, 179–185. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science (80-.) 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ye, J.; Dong, Z.; Hu, S.; Xiao, M. Novel genetic alterations and their impact on target therapy response in head and neck squamous cell carcinoma. Cancer Manag. Res. 2019, 11, 1321. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science (80-.) 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.N.; Van Waes, C.; Seiwert, T.Y. Genetic landscape of human papillomavirus-associated head and neck cancer and comparison to tobacco-related tumors. J. Clin. Oncol. 2015, 33, 3227–3234. [Google Scholar] [CrossRef]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef]
- Revathidevi, S.; Murugan, A.K.; Nakaoka, H.; Inoue, I.; Munirajan, A.K. APOBEC: A molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021, 496, 104–116. [Google Scholar] [CrossRef]
- Siriwardena, S.U.; Chen, K.; Bhagwat, A.S. Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases. Chem. Rev. 2016, 116, 12688–12710. [Google Scholar] [CrossRef]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Betts, L.; Xiang, S.; Short, S.A.; Wolfenden, R.; Carter, C.W. Cytidine deaminase. the 2.3 Å crystal structure of an enzyme: Transition-state analog complex. J. Mol. Biol. 1994, 235, 635–656. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Li, M.; Carpenter, M.A.; McDougle, R.M.; Luengas, E.M.; Macdonald, P.J.; Harris, R.S.; Mueller, J.D. APOBEC3 multimerization correlates with HIV-1 packaging and restriction activity in living cells. J. Mol. Biol. 2014, 426, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Sacho, E.J.; Tessmer, I.; Croteau, D.L.; Erie, D.A.; Diaz, M. Activation-induced deaminase, AID, is catalytically active as a monomer on single-stranded DNA. DNA Repair (Amst) 2008, 7, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, N.; Sarwar, R. An overview of cytidine deaminases. Int. J. Hematol. 2006, 83, 195–200. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of viral infections by innate immunity. Biochem. Pharmacol. 2020, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.J.; Fenton, T.R. The APOBEC3 genes and their role in cancer: Insights from human papillomavirus. J. Mol. Endocrinol. 2019, 62, R269–R287. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Ross, S.R. APOBEC3 Proteins in Viral Immunity. J. Immunol. 2015, 195, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009, 5, e1000439. [Google Scholar] [CrossRef]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of Hepatitis B Virus Replication by APOBEC3G. Science (80-.) 2004, 303, 1829. [Google Scholar] [CrossRef]
- Suspène, R.; Guétard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef]
- Peretti, A.; Geoghegan, E.M.; Pastrana, D.V.; Smola, S.; Feld, P.; Sauter, M.; Lohse, S.; Ramesh, M.; Lim, E.S.; Wang, D.; et al. Characterization of BK Polyomaviruses from Kidney Transplant Recipients Suggests a Role for APOBEC3 in Driving In-Host Virus Evolution. Cell Host Microbe 2018, 23, 628–635.e7. [Google Scholar] [CrossRef]
- Verhalen, B.; Starrett, G.J.; Harris, R.S.; Jiang, M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J. Virol. 2016, 90, 6379–6386. [Google Scholar] [CrossRef]
- Suspene, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic Editing of Herpes Simplex Virus 1 and Epstein-Barr Herpesvirus Genomes by Human APOBEC3 Cytidine Deaminases in Culture and In Vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Oiknine-Djian, E.; Zakay-Rones, Z.; Vorontsov, O.; Haimov-Kochman, R.; Nevo, Y.; Stockheim, D.; Yagel, S.; Panet, A.; Wolf, D.G. APOBEC3A Is Upregulated by Human Cytomegalovirus (HCMV) in the Maternal-Fetal Interface, Acting as an Innate Anti-HCMV Effector. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.Z.; Yockteng-Melgar, J.; Jarvis, M.C.; Malik-Soni, N.; Borozan, I.; Carpenter, M.A.; McCann, J.L.; Ebrahimi, D.; Shaban, N.M.; Marcon, E.; et al. Epstein–Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol. 2019, 4, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.Z.; Moraes, S.N.; Attarian, C.; Yockteng-Melgar, J.; Jarvis, M.C.; Biolatti, M.; Galitska, G.; Dell’Oste, V.; Frappier, L.; Bierle, C.J.; et al. A Conserved Mechanism of APOBEC3 Relocalization by Herpesviral Ribonucleotide Reductase Large Subunits. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Westrich, J.A.; Van Doorslaer, K.; Pyeon, D. Roles of APOBEC3A and APOBEC3B in human papillomavirus infection and disease progression. Viruses 2017, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Lambert, P.F.; Ahlquist, P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 9311–9316. [Google Scholar] [CrossRef]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A Functions as a Restriction Factor of Human Papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Thomas Taggart, R.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef]
- Land, A.M.; Law, E.K.; Carpenter, M.A.; Lackey, L.; Brown, W.L.; Harris, R.S. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J. Biol. Chem. 2013, 288, 17253–17260. [Google Scholar] [CrossRef] [PubMed]
- Mussil, B.; Suspène, R.; Aynaud, M.M.; Gauvrit, A.; Vartanian, J.P.; Wain-Hobson, S. Human APOBEC3A Isoforms Translocate to the Nucleus and Induce DNA Double Strand Breaks Leading to Cell Stress and Death. PLoS ONE 2013, 8, e73641. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A.; Browne, H.M.; Appleby, M.; Minson, A.C. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int. J. Cancer 1989, 43, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto, I.; Mori, S.; Aoyama, S.; Wakae, K.; Muramatsu, M.; Kondo, K. Hypermutation in the E2 gene of human papillomavirus type 16 in cervical intraepithelial neoplasia. J. Med. Virol. 2015, 87, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Guétard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science (80-.) 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 Deaminases Induce Hypermutation in Human Papillomavirus 16 DNA upon Beta Interferon Stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef]
- Hirose, Y.; Onuki, M.; Tenjimbayashi, Y.; Mori, S.; Ishii, Y.; Takeuchi, T.; Tasaka, N.; Satoh, T.; Morisada, T.; Iwata, T.; et al. Within-Host Variations of Human Papillomavirus Reveal APOBEC Signature Mutagenesis in the Viral Genome. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6. [Google Scholar] [CrossRef]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Guo, K.; Liu, C.-W.; Santiago, M.L.; Pyeon, D. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Henderson, S.; Thirdborough, S.M.; Ottensmeier, C.H.; Su, X.; Lechner, M.; Feber, A.; Thomas, G.J.; Fenton, T.R. Human papillomavirus drives tumor development throughout the head and neck: Improved prognosis is associated with an immune response largely restricted to the Oropharynx. J. Clin. Oncol. 2016, 34, 4132–4141. [Google Scholar] [CrossRef]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Yugawa, T.; Kiyono, T.; Nishina, H.; Kukimoto, I. Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Kukimoto, I. Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem. Biophys. Res. Commun. 2015, 460, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Steiner, L.; Engeland, K. The transcription factor p53: Not a repressor, solely an activator. Cell Cycle 2014, 13, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, M.; Singh, A.K.; Gemma, C.; Kranjec, C.; Farzan, R.; Leach, D.A.; Navaratnam, N.; Pálinkás, H.L.; Vertessy, B.G.; Fenton, T.R.; et al. P53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res. 2017, 45, 11056–11069. [Google Scholar] [CrossRef] [PubMed]
- Cannataro, V.L.; Gaffney, S.G.; Sasaki, T.; Issaeva, N.; Grewal, N.K.S.; Grandis, J.R.; Yarbrough, W.G.; Burtness, B.; Anderson, K.S.; Townsend, J.P. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene 2019, 38, 3475–3487. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Zhang, Y.; Zarins, K.R.; Jones, T.R.; Virani, S.; Peterson, L.A.; McHugh, J.B.; Chepeha, D.; Wolf, G.T.; Rozek, L.S.; et al. Expressed HNSCC variants by HPV-status in a well-characterized Michigan cohort. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef]
- Kondo, S.; Wakae, K.; Wakisaka, N.; Nakanishi, Y.; Ishikawa, K.; Komori, T.; Moriyama-Kita, M.; Endo, K.; Murono, S.; Wang, Z.; et al. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene 2017, 36, 1687–1697. [Google Scholar] [CrossRef]
- Chatfield-Reed, K.; Gui, S.; O’Neill, W.Q.; Teknos, T.N.; Pan, Q. HPV33+ HNSCC is associated with poor prognosis and has unique genomic and immunologic landscapes. Oral Oncol. 2020, 100, 104488. [Google Scholar] [CrossRef]
- Kono, T.; Hoover, P.; Poropatich, K.; Paunesku, T.; Mittal, B.B.; Samant, S.; Laimins, L.A. Activation of DNA damage repair factors in HPV positive oropharyngeal cancers. Virology 2020, 547, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, Y.W.; Cao, R.Y.; Pan, X.; Chen, X.J.; Zhang, S.Y.; Zhang, W.L.; Zhou, J.Y.; Cheng, B.; Ren, X.Y. Association between APOBEC3H-Mediated Demethylation and Immune Landscape in Head and Neck Squamous Carcinoma. Biomed. Res. Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Lee, C.C.; Liu, H.; Wu, C.S.; Pickering, C.R.; Huang, P.J.; Wang, J.; Chang, I.Y.F.; Yeh, Y.M.; Chen, C.D.; et al. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Alexandrov, L.B.; Den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Argyris, P.P.; Wilkinson, P.E.; Jarvis, M.C.; Magliocca, K.R.; Patel, M.R.; Vogel, R.I.; Gopalakrishnan, R.; Koutlas, I.G.; Harris, R.S. Endogenous APOBEC3B overexpression characterizes HPV-positive and HPV-negative oral epithelial dysplasias and head and neck cancers. Mod. Pathol. 2020. [Google Scholar] [CrossRef]
- Fanourakis, G.; Tosios, K.; Papanikolaou, N.; Chatzistamou, I.; Xydous, M.; Tseleni-Balafouta, S.; Sklavounou, A.; Voutsinas, G.E.; Vastardis, H. Evidence for APOBEC3B mRNA and protein expression in oral squamous cell carcinomas. Exp. Mol. Pathol. 2016, 101, 314–319. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Kondo, S.; Wakisaka, N.; Tsuji, A.; Endo, K.; Murono, S.; Ito, M.; Kitamura, K.; Muramatsu, M.; Yoshizaki, T. Role of Activation-Induced Cytidine Deaminase in the Development of Oral Squamous Cell Carcinoma. PLoS ONE 2013, 8, e62066. [Google Scholar] [CrossRef]
- Kano, M.; Kondo, S.; Wakisaka, N.; Wakae, K.; Aga, M.; Moriyama-Kita, M.; Ishikawa, K.; Ueno, T.; Nakanishi, Y.; Hatano, M.; et al. Expression of estrogen receptor alpha is associated with pathogenesis and prognosis of human papillomavirus-positive oropharyngeal cancer. Int. J. Cancer 2019, 145, 1547–1557. [Google Scholar] [CrossRef]
- Messerschmidt, C.; Obermayer, B.; Klinghammer, K.; Ochsenreither, S.; Treue, D.; Stenzinger, A.; Glimm, H.; Fröhling, S.; Kindler, T.; Brandts, C.H.; et al. Distinct immune evasion in APOBEC-enriched, HPV-negative HNSCC. Int. J. Cancer 2020, 147, 2293–2302. [Google Scholar] [CrossRef]
- Faden, D.L.; Ding, F.; Lin, Y.; Zhai, S.; Kuo, F.; Chan, T.A.; Morris, L.G.; Ferris, R.L. APOBEC mutagenesis is tightly linked to the immune landscape and immunotherapy biomarkers in head and neck squamous cell carcinoma. Oral Oncol. 2019, 96, 140–147. [Google Scholar] [CrossRef]
- Conner, K.L.; Shaik, A.N.; Ekinci, E.; Kim, S.; Ruterbusch, J.J.; Cote, M.L.; Patrick, S.M. HPV induction of APOBEC3 enzymes mediate overall survival and response to cisplatin in head and neck cancer. DNA Repair (Amst) 2020, 87, 102802. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| HPV+ | HPV- |
|---|---|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riva, G.; Albano, C.; Gugliesi, F.; Pasquero, S.; Pacheco, S.F.C.; Pecorari, G.; Landolfo, S.; Biolatti, M.; Dell’Oste, V. HPV Meets APOBEC: New Players in Head and Neck Cancer. Int. J. Mol. Sci. 2021, 22, 1402. https://doi.org/10.3390/ijms22031402
Riva G, Albano C, Gugliesi F, Pasquero S, Pacheco SFC, Pecorari G, Landolfo S, Biolatti M, Dell’Oste V. HPV Meets APOBEC: New Players in Head and Neck Cancer. International Journal of Molecular Sciences. 2021; 22(3):1402. https://doi.org/10.3390/ijms22031402
Chicago/Turabian StyleRiva, Giuseppe, Camilla Albano, Francesca Gugliesi, Selina Pasquero, Sergio Fernando Castillo Pacheco, Giancarlo Pecorari, Santo Landolfo, Matteo Biolatti, and Valentina Dell’Oste. 2021. "HPV Meets APOBEC: New Players in Head and Neck Cancer" International Journal of Molecular Sciences 22, no. 3: 1402. https://doi.org/10.3390/ijms22031402
APA StyleRiva, G., Albano, C., Gugliesi, F., Pasquero, S., Pacheco, S. F. C., Pecorari, G., Landolfo, S., Biolatti, M., & Dell’Oste, V. (2021). HPV Meets APOBEC: New Players in Head and Neck Cancer. International Journal of Molecular Sciences, 22(3), 1402. https://doi.org/10.3390/ijms22031402