
Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
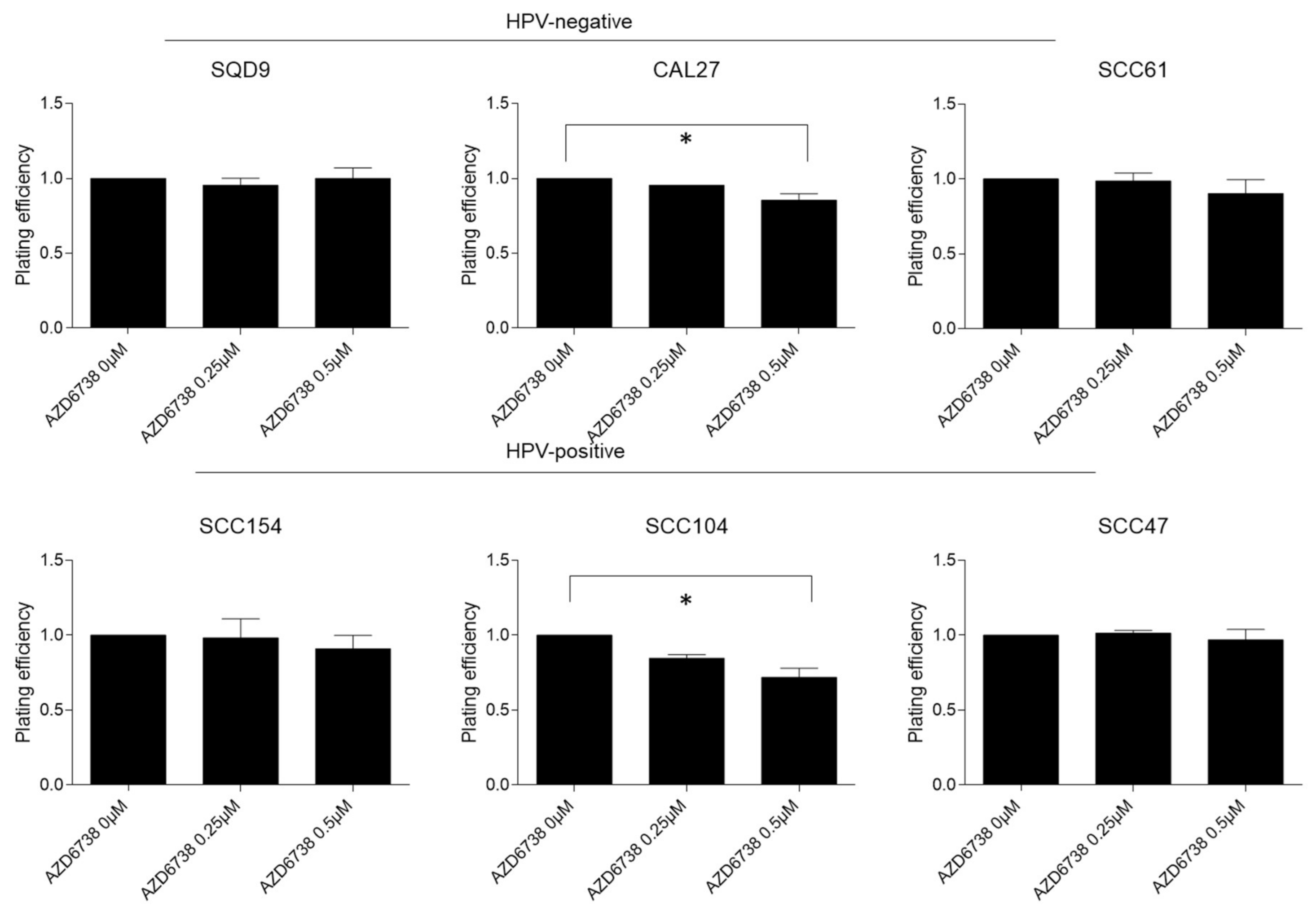
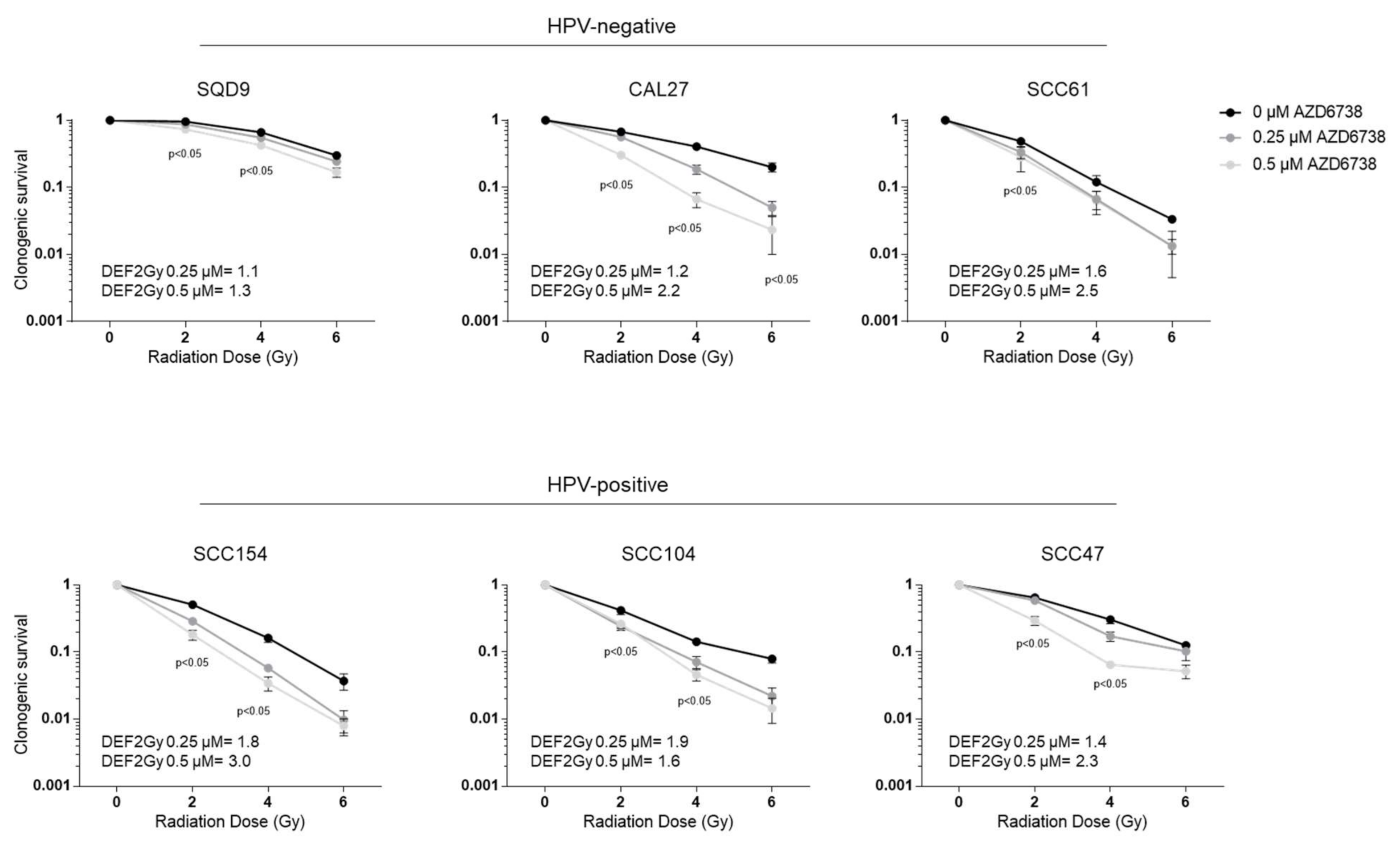
2.1. ATR Inhibition Induces Radiosensitization of HPV-Negative and HPV-Positive HNSCC In Vitro
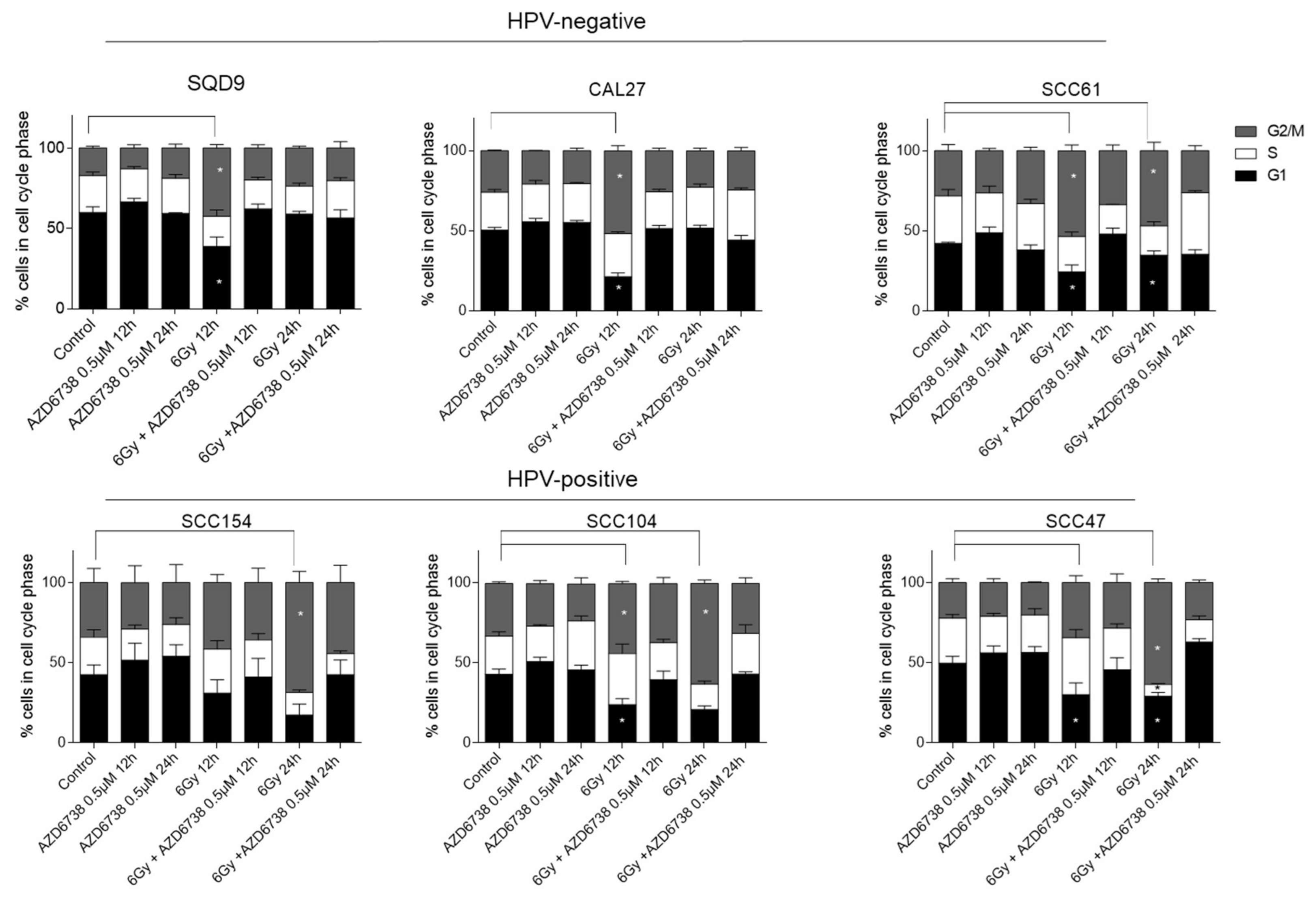
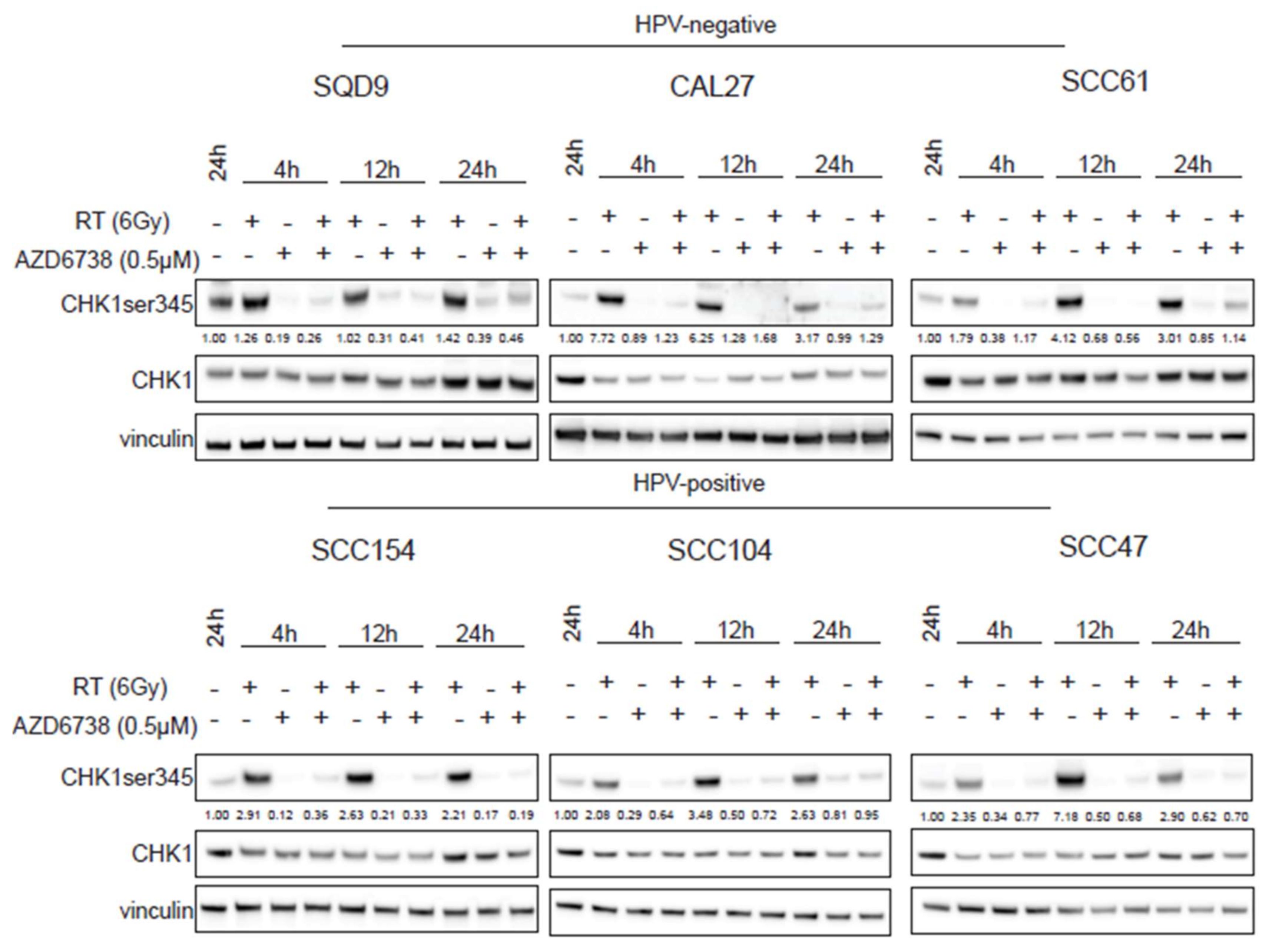
2.2. ATR Induces Abrogation of Cell Cycle Checkpoint by Inhibiting CHK1 Phosphorylation
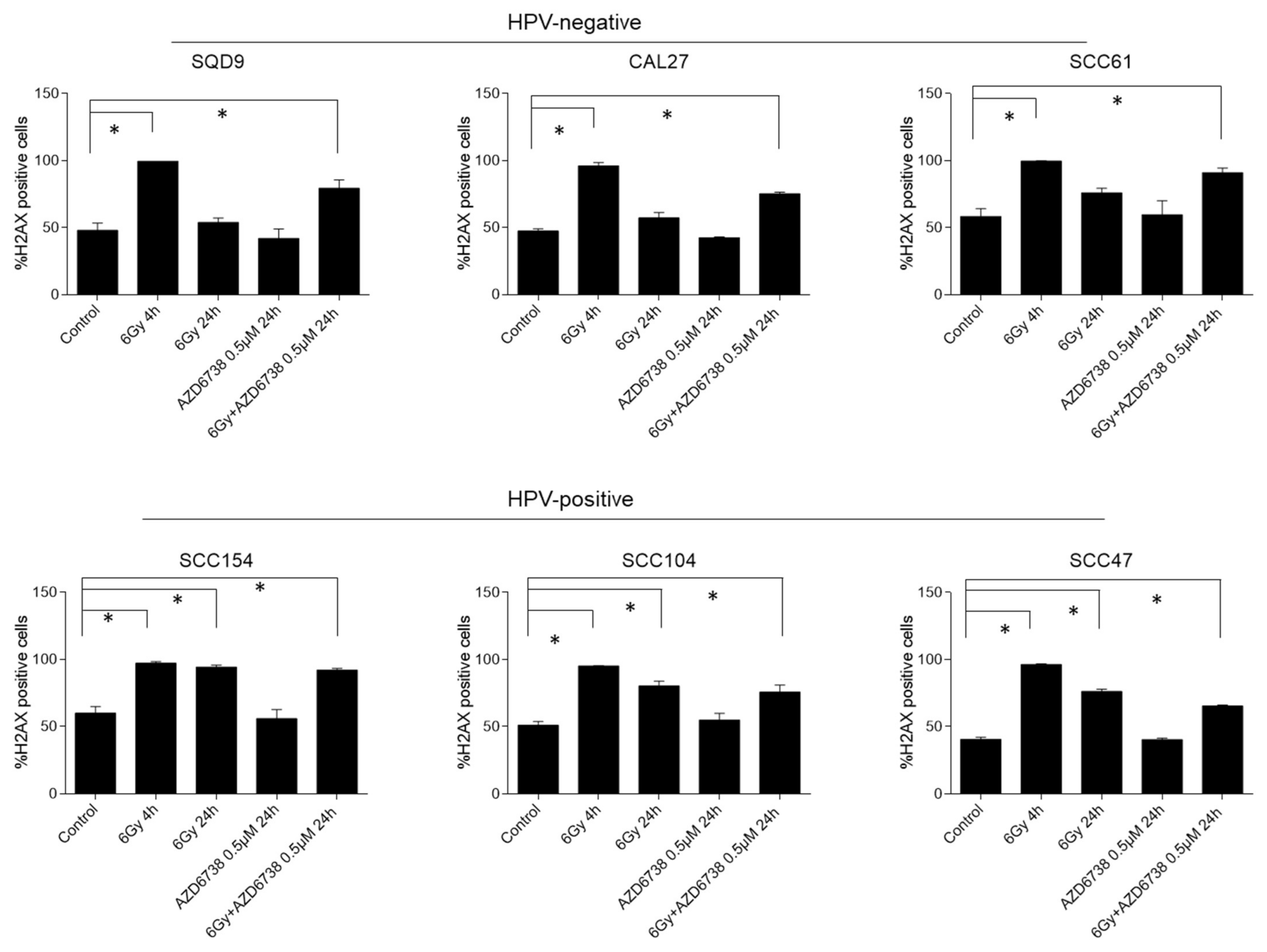
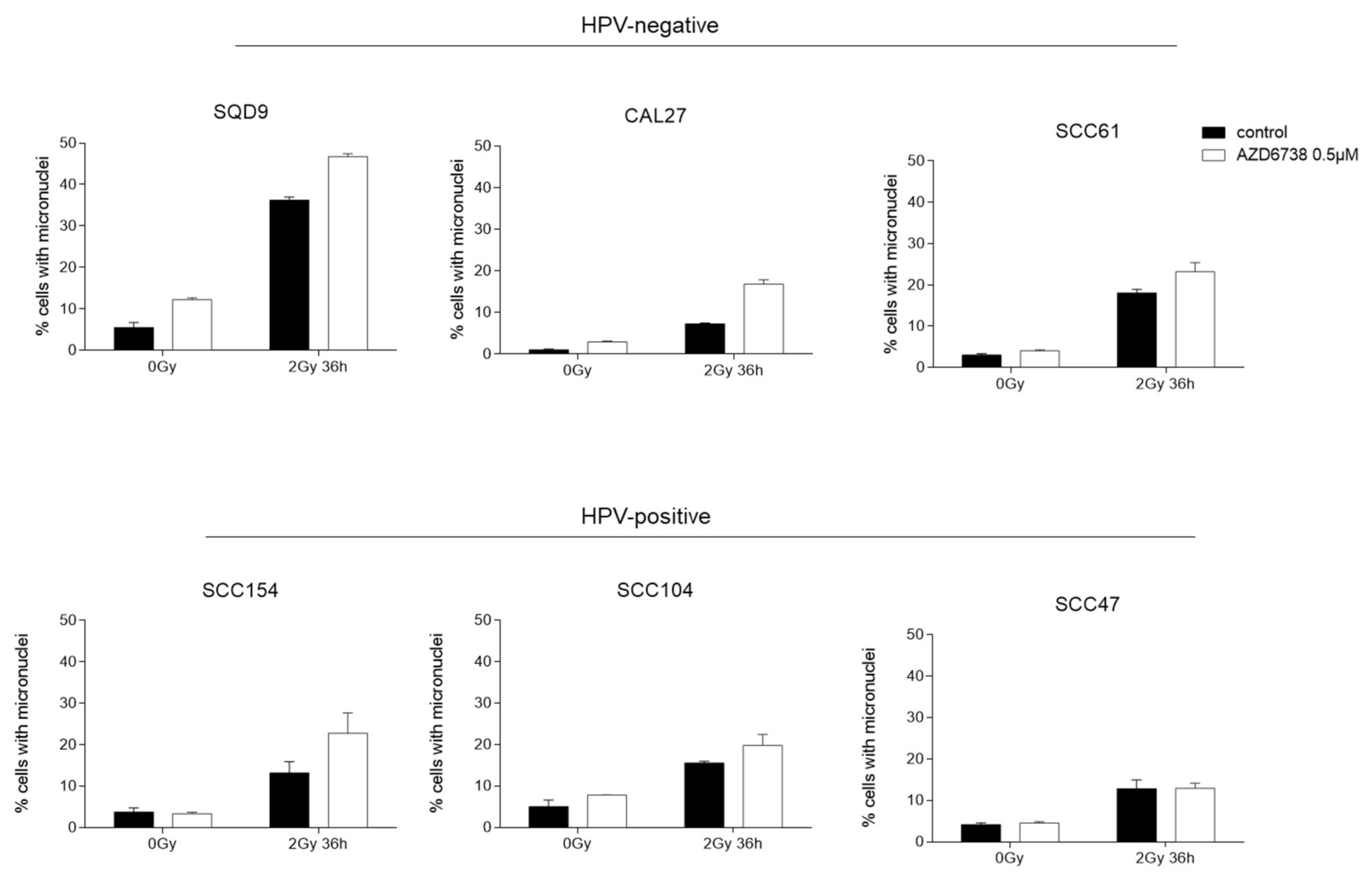
2.3. ATR Inhibition Induces an Increase in Residual γH2AX Levels and Micronuclei in HNSCC Cells
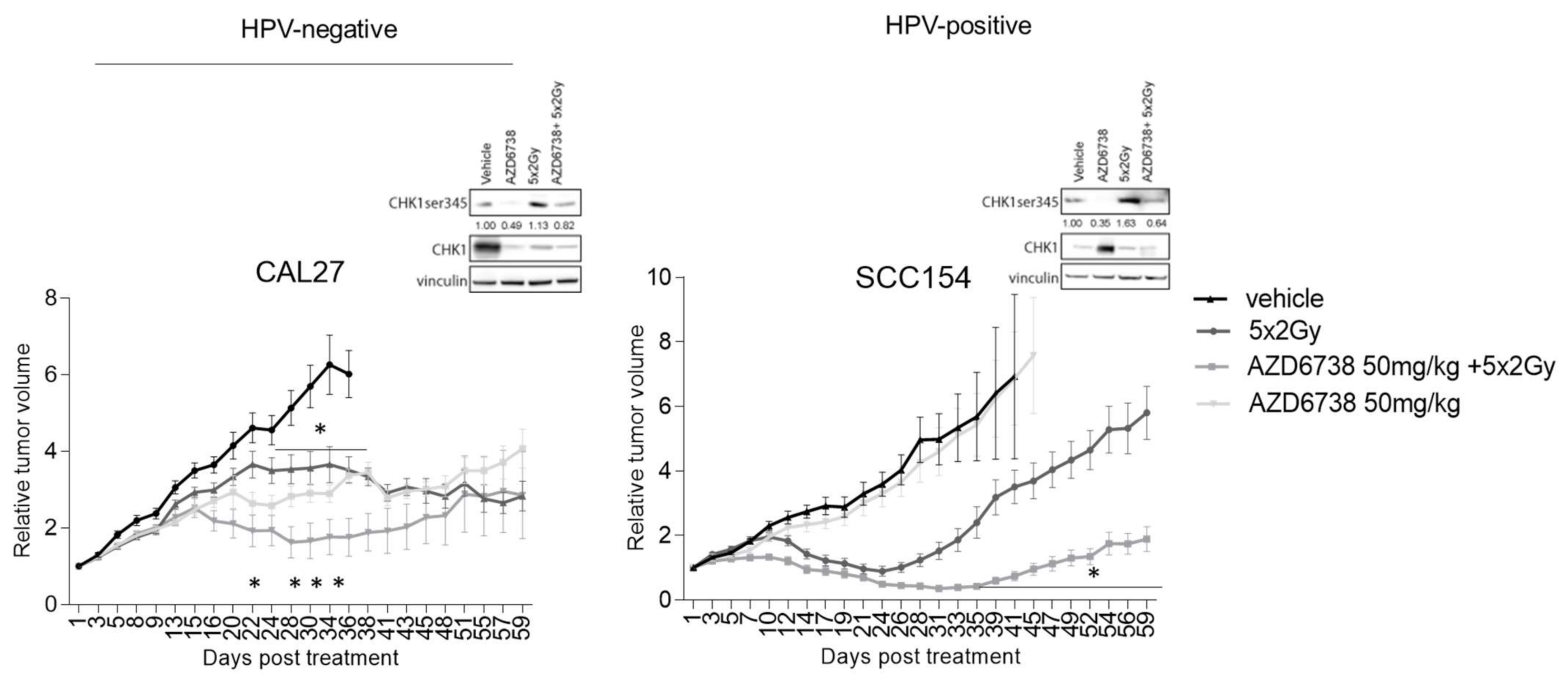
2.4. Preclinical Assessment of ATR Inhibitor AZD6738 as Radiosensitizer for HPV-Positive and HPV-Negative HNSCC
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Consumables
4.2. Colony Assay
4.3. Western Blotting
4.4. Flow Cytometry
4.5. Micronuclei Assay
4.6. Xenograft Models
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Amelio, I.; Urbano, T.G.; Tavassoli, M. Clinical update on cancer: Molecular oncology of head and neck cancer. Cell Death Dis. 2014, 5, e1018. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.-K.N.D.; Mierzwa, M.; D’Silva, N.J. Radiation resistance in head and neck squamous cell carcinoma: Dire need for an appropriate sensitizer. Oncogene 2020, 39, 3638–3649. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Bamps, M.; Glorieux, M.; Zhao, P.; Sablina, A.A.; Nuyts, S. Radiosensitization approaches for HPV-positive and HPV-negative head and neck squamous carcinomas. Int. J. Cancer 2020, 146, 1075–1085. [Google Scholar] [CrossRef]
- Glorieux, M.; Dok, R.; Nuyts, S. Novel DNA targeted therapies for head and neck cancers: Clinical potential and biomarkers. Oncotarget 2017, 8, 81662–81678. [Google Scholar] [CrossRef]
- Velic, D.; Couturier, A.M.; Ferreira, M.T.; Rodrigue, A.; Poirier, G.G.; Fleury, F.; Masson, J.-Y. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomology 2015, 5, 3204–3259. [Google Scholar] [CrossRef]
- Massagué, J. G1 cell-cycle control and cancer. Nat. Cell Biol. 2004, 432, 298–306. [Google Scholar] [CrossRef]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25–34. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.M.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2016, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Goetz, M.P.; et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 1–27. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Arenz, R.N.A.; Ziemann, F.; Mayer, C.; Wittig, A.; Dreffke, K.; Preising, S.; Wagner, S.; Klussmann, J.-P.; Engenhart-Cabillic, R.; Wittekindt, C. Increased radiosensitivity of HPV-positive head and neck cancer cell lines due to cell cycle dysregulation and induction of apoptosis. Strahlenther. und Onkol. 2014, 190, 839–846. [Google Scholar] [CrossRef]
- Dok, R.; Kalev, P.; Van Limbergen, E.J.; Asbagh, L.A.; Vázquez, I.; Hauben, E.; Sablina, A.; Nuyts, S. p16INK4a Impairs Homologous Recombination–Mediated DNA Repair in Human Papillomavirus–Positive Head and Neck Tumors. Cancer Res. 2014, 74, 1739–1751. [Google Scholar] [CrossRef]
- Dok, R.; Nuyts, S. HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers 2016, 8, 41. [Google Scholar] [CrossRef]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced Radiation Sensitivity in HPV-Positive Head and Neck Cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef]
- Nickson, C.M.; Moori, P.; Carter, R.J.; Rubbi, C.P.; Parsons, J.L. Misregulation of DNA damage repair pathways in HPV-positive head and neck squamous cell carcinoma contributes to cellular radiosensitivity. Oncotarget 2017, 8, 29963–29975. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Nickel, K.P.; Torres, A.D.; Lee, D.; Lambert, P.F.; Kimple, R.J. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother. Oncol. 2014, 113, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, T.; Tribius, S.; Grob, T.; Meyer, F.; Busch, C.-J.; Petersen, C.; Dikomey, E.; Kriegs, M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol. 2013, 107, 242–246. [Google Scholar] [CrossRef]
- Spriggs, C.C.; Laimins, L. Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication. Viruses 2017, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, P.-J.; Molkentine, D.P.; Chen, C.; Molkentine, J.M.; Piao, H.; Raju, U.; Zhang, J.; Valdecanas, D.R.; Tailor, R.C.; et al. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene 2016, 36, 820–828. [Google Scholar] [CrossRef]
- Benada, J.; Macurek, L. Targeting the Checkpoint to Kill Cancer Cells. Biomol. 2015, 5, 1912–1937. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Brown, E.J. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003, 17, 615–628. [Google Scholar] [CrossRef]
- Weber, A.M.; Ryan, A. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Moore, D.; Parker, C.J.; Broker, T.R.; Chow, L.T. Targeting DNA Damage Response as a Strategy to Treat HPV Infections. Int. J. Mol. Sci. 2019, 20, 5455. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Moody, C.A. Impact of the DNA Damage Response on Human Papillomavirus Chromatin. PLOS Pathog. 2016, 12, e1005613. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Parsons, J.L. The radiobiology of HPV-positive and HPV-negative head and neck squamous cell carcinoma. Expert Rev. Mol. Med. 2020, 22, e3. [Google Scholar] [CrossRef]
- Vitti, E.T.; Kacperek, A.; Parsons, J.L. Targeting DNA Double-Strand Break Repair Enhances Radiosensitivity of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma to Photons and Protons. Cancers 2020, 12, 1490. [Google Scholar] [CrossRef]
- Leonard, B.C.; Lee, E.D.; Bhola, N.E.; Li, H.; Sogaard, K.K.; Bakkenist, C.J.; Grandis, J.R.; Johnson, D.E. ATR inhibition sensitizes HPV− and HPV+ head and neck squamous cell carcinoma to cisplatin. Oral Oncol. 2019, 95, 35–42. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Feeney, L.; Revet, I. Phosphorylated H2Ax is not an unambiguous marker for DNA double-strand breaks. Cell Cycle 2011, 10, 3223–3224. [Google Scholar] [CrossRef]
- Reddig, A.; Roggenbuck, D.; Reinhold, D. Comparison of different immunoassays for γH2AX quantification. J. Lab. Precis. Med. 2018, 3, 80. [Google Scholar] [CrossRef]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; LaLonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef]
- Dillon, M.; Guevara, J.; Mohammed, K.; Smith, S.; Dean, E.; McLellan, L.; Boylan, Z.; Spicer, J.; Forster, M.; Harrington, K. A phase I study of ATR inhibitor, AZD6738, as monotherapy in advanced solid tumours (PATRIOT part A, B). Ann. Oncol. 2019, 30, v165–v166. [Google Scholar] [CrossRef]
- Hafsi, H.; Dillon, M.T.; Barker, H.E.; Kyula, J.N.; Schick, U.; Paget, J.T.; Smith, H.G.; Pedersen, M.; McLaughlin, M.; Harrington, K.J. Combined ATR and DNA-PK Inhibition Radiosensitizes Tumor Cells Independently of Their p53 Status. Front. Oncol. 2018, 8, 245. [Google Scholar] [CrossRef]
- Glorieux, M.; Dok, R.; Nuyts, S. The influence of PI3K inhibition on the radiotherapy response of head and neck cancer cells. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dok, R.; Glorieux, M.; Bamps, M.; Nuyts, S. Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers. Int. J. Mol. Sci. 2021, 22, 1504. https://doi.org/10.3390/ijms22041504
Dok R, Glorieux M, Bamps M, Nuyts S. Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers. International Journal of Molecular Sciences. 2021; 22(4):1504. https://doi.org/10.3390/ijms22041504
Chicago/Turabian StyleDok, Rüveyda, Mary Glorieux, Marieke Bamps, and Sandra Nuyts. 2021. "Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers" International Journal of Molecular Sciences 22, no. 4: 1504. https://doi.org/10.3390/ijms22041504
APA StyleDok, R., Glorieux, M., Bamps, M., & Nuyts, S. (2021). Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers. International Journal of Molecular Sciences, 22(4), 1504. https://doi.org/10.3390/ijms22041504