
Insulin’s Discovery: New Insights on Its Hundredth Birthday: From Insulin Action and Clearance to Sweet Networks


Abstract
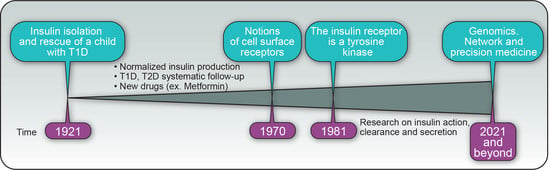
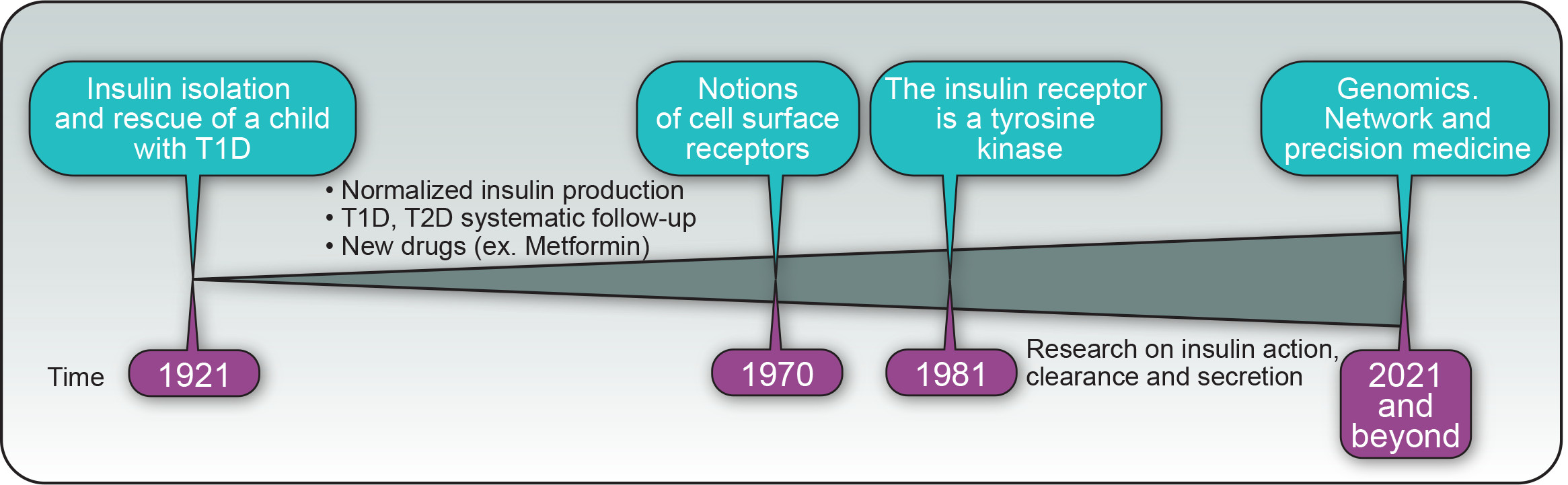{kind=link}
{kind=link}
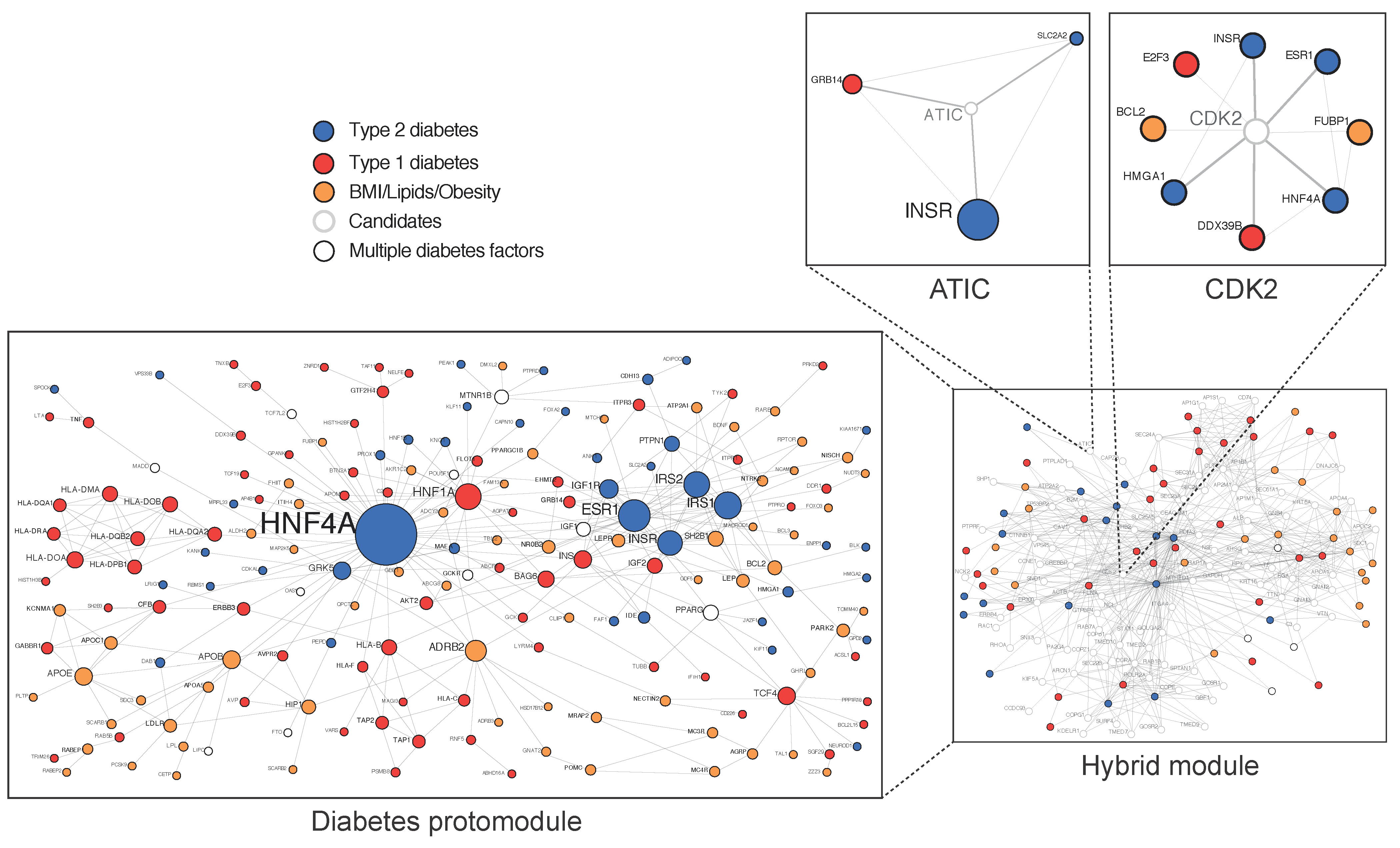{kind=link}
{kind=link}
1. Introduction
2. The Insulin Receptor-Tyrosine Kinase (IR)
3. IR Endocytosis and Insulin Clearance
4. From PPIN to a T2D Disease Module
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barron, M. The relation of the islets of Langerhans with special reference to cases of pancreates lithiasis. Sur. Gynecol. Obstet. 1920, 31, 437–448. [Google Scholar]
- MacLeod, J.J.R. Insulin and the steps taken to secure an effective preparation. Can. Med. Assoc. J. 1922, 12, 899–900. [Google Scholar]
- Noble, R.L. Memories of james bertram collip. Can. Med. Assoc. J. 1965, 93, 1356–1364. [Google Scholar]
- Bliss, M. Resurrections in Toronto: The emergence of insulin. Horm. Res. Paediatr. 2005, 64, 98–102. [Google Scholar] [CrossRef]
- Bliss, M. The eclipse and rehabilitation of JJR Macleod, Scotland’s insulin laureate. J. R. Coll. Physicians Edinb. 2013, 43, 366–373. [Google Scholar] [CrossRef]
- Joslin, E.P. The use of insulin in its various forms in the treatment of diabetes. Bull. N. Y. Acad. Med. 1942, 18, 200–216. [Google Scholar]
- Berghe, G.V.D.; Wouters, P.J.; Bouillon, R.; Weekers, F.; Verwaest, C.; Schetz, M.; Vlasselaers, D.; Ferdinande, P.; Lauwers, P. Outcome benefit of intensive insulin therapy in the critically ill: Insulin dose versus glycemic control. Crit. Care Med. 2003, 31, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L.E. IDF diabetes atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. The target of metformin in type 2 diabetes. N. Engl. J. Med. 2014, 371, 1547–1548. [Google Scholar] [CrossRef]
- Feig, D.S. Preventing diabetes in women with gestational diabetes. Diabetes/Metab. Res. Rev. 2012, 28, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Kaprio, J.; Tuomilehto, J.; Koskenvuo, M.; Romanov, K.; Reunanen, A.; Eriksson, J.; Stengård, J.; Kesäniemi, Y.A. Concordance for Type 1 (insulin-dependent) and Type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992, 35, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.; Kyvik, K.O.; Vaag, A.; Beck-Nielsen, H. Heritability of Type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—A population-based twin study. Diabetologia 1999, 42, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Zaitlen, N.; Hu, F.B.; Kraft, P.; Price, A.L. Genetic and environmental components of family history in type 2 diabetes. Qual. Life Res. 2014, 134, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nat. Cell Biol. 2016, 536, 41–47. [Google Scholar] [CrossRef]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An expanded genome-wide association study of Type 2 diabetes in Europeans. Diabetes 2017, 66, 2888–2902. [Google Scholar] [CrossRef]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.; et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Landry, C.R.; Levy, E.D.; Abd-Rabbo, D.; Tarassov, K.; Michnick, S.W. Extracting insight from noisy cellular networks. Cell 2013, 155, 983–989. [Google Scholar] [CrossRef]
- Choobdar, S.; Ahsen, M.E.; Crawford, J.; Tomasoni, M.; Fang, T.; Lamparter, D.; Lin, J.; Hescott, B.; Hu, X.; Mercer, J.; et al. Assessment of network module identification across complex diseases. Nat. Methods 2019, 16, 843–852. [Google Scholar] [CrossRef]
- Cafarelli, T.; Desbuleux, A.; Wang, Y.; Choi, S.G.; De Ridder, D.; Vidal, M. Mapping, modeling, and characterization of protein–protein interactions on a proteomic scale. Curr. Opin. Struct. Biol. 2017, 44, 201–210. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef]
- Boutchueng-Djidjou, M.; Belleau, P.; Bilodeau, N.; Fortier, S.; Bourassa, S.; Droit, A.; Elowe, S.; Faure, R.L. A type 2 diabetes disease module with a high collective influence for Cdk2 and PTPLAD1 is localized in endosomes. PLoS ONE 2018, 13, e0205180. [Google Scholar] [CrossRef] [PubMed]
- Estrada, K.; Aukrust, I.; Bjørkhaug, L.; Burtt, N.P.; Mercader, J.M.; García-Ortiz, H.; Huerta-Chagoya, A.; Moreno-Macías, H.; Walford, G.; Flannick, J.; et al. Association of a low-frequency variant inHNF1AWith Type 2 diabetes in a latino population. JAMA 2014, 311, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, J.J.R. The problem of the fundamental action of insulin. Can. Med. Assoc. J. 1925, 15, 476–478. [Google Scholar]
- Lefkowitz, R.J.; Roth, J.; Pastan, I. Radioreceptor assay of adrenocorticotropic hormone: New approach to assay of polypeptide hormones in plasma. Science 1970, 170, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Freychet, P.; Roth, J.; Neville, D.M. Insulin receptors in the liver: Specific binding of [125I]insulin to the plasma membrane and its relation to insulin bioactivity. Proc. Natl. Acad. Sci. USA 1971, 68, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Sefton, B.M. Transforming gene product of rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. USA 1980, 77, 1311–1315. [Google Scholar] [CrossRef]
- Kasuga, M.; Zick, Y.; Blithe, D.L.; Crettaz, M.; Kahn, C.R. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nat. Cell Biol. 1982, 298, 667–669. [Google Scholar] [CrossRef]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.-H.; Masiarz, F.; Kan, Y.; Goldfine, I.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.-C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nat. Cell Biol. 1985, 313, 756–761. [Google Scholar] [CrossRef]
- Kadowaki, T.; Bevins, C.L.; Cama, A.; Ojamaa, K.; Marcus-Samuels, B.; Beitz, L.; McKeon, C.; Taylor, S.I. Two mutant alleles of the insulin receptor gene in a patient with extreme insulin resistance. Science 1988, 240, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Scapin, G.; Dandey, V.P.; Zhang, Z.; Prosise, W.; Hruza, A.; Kelly, T.; Mayhood, T.; Strickland, C.; Potter, C.S.; Carragher, B. Structure of the insulin receptor–insulin complex by single-particle cryo-EM analysis. Nat. Cell Biol. 2018, 556, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, T.; Schäfer, I.B.; Poojari, C.; Brankatschk, B.; Vattulainen, I.; Strauss, M.; Coskun, Ü. Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain. J. Cell Biol. 2020, 219, 1. [Google Scholar] [CrossRef]
- Uchikawa, E.; Choi, E.; Shang, G.; Yu, H.; Bai, X. Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor–ligand complex. eLife 2019, 8, e48630. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform a, a newly recognized, high-affinity insulin-like growth factor ii receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- McClain, D.A. Mechanism and role of insulin receptor endocytosis. Am. J. Med. Sci. 1992, 304, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Terris, S.; Steiner, D.F. Binding and degradation of 125I-insulin by rat hepatocytes. J. Biol. Chem. 1975, 250, 8389–8398. [Google Scholar] [CrossRef]
- Heinrich, G.; Ghadieh, H.E.; Ghanem, S.S.; Muturi, H.T.; Rezaei, K.; Al-Share, Q.Y.; Bowman, T.A.; Zhang, D.; Garofalo, R.S.; Yin, L.; et al. Loss of hepatic CEACAM1: A unifying mechanism linking insulin resistance to obesity and non-alcoholic fatty liver disease. Front. Endocrinol. 2017, 8, 8. [Google Scholar] [CrossRef]
- Najjar, S.M. Regulation of insulin action by CEACAM1. Trends Endocrinol. Metab. 2002, 13, 240–245. [Google Scholar] [CrossRef]
- Poy, M.N.; Yang, Y.; Rezaei, K.; Fernström, M.A.; Lee, A.D.; Kido, Y.; Erickson, S.K.; Najjar, S.M. CEACAM1 regulates insulin clearance in liver. Nat. Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef]
- Mirsky, I.A.; Broh-Kahn, R.H. The inactivation of insulin by tissue extracts; the distribution and properties of insulin inactivating extracts. Arch. Biochem. 1949, 20, 1–9. [Google Scholar]
- Shii, K.; Yokono, K.; Baba, S.; Roth, R.A. Purification and characterization of insulin-degrading enzyme from human erythrocytes. Diabetes 1986, 35, 675–683. [Google Scholar] [CrossRef]
- Shii, K.; Baba, S.; Yokono, K.; Roth, R.A. Covalent linkage of 125I-insulin to a cytosolic insulin-degrading enzyme. J. Biol. Chem. 1985, 260, 6503–6506. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin degradation: Progress and potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar]
- Duckworth, W.C.; Hamel, F.G.; Peavy, D.E. Hepatic metabolism of insulin. Am. J. Med. 1988, 85, 71–76. [Google Scholar] [CrossRef]
- Collinet, C.; Stöter, M.; Bradshaw, C.R.; Samusik, N.; Rink, J.C.; Kenski, D.M.; Habermann, B.; Buchholz, F.; Henschel, R.; Mueller, M.S.; et al. Systems survey of endocytosis by multiparametric image analysis. Nat. Cell Biol. 2010, 464, 243–249. [Google Scholar] [CrossRef]
- Zeigerer, A.; Bogorad, R.L.; Sharma, K.; Gilleron, J.; Seifert, S.; Sales, S.; Berndt, N.; Bulik, S.; Marsico, G.; D’Souza, R.C.J.; et al. Regulation of liver metabolism by the endosomal GTPase Rab5. Cell Rep. 2015, 11, 884–892. [Google Scholar] [CrossRef]
- Choi, E.; Zhang, X.; Xing, C.; Yu, H. Mitotic checkpoint regulators control insulin signaling and metabolic homeostasis. Cell 2016, 166, 567–581. [Google Scholar] [CrossRef]
- Choi, E.; Yu, H. Spindle checkpoint regulators in insulin signaling. Front. Cell Dev. Biol. 2018, 6, 161. [Google Scholar] [CrossRef]
- Choi, E.; Kikuchi, S.; Gao, H.; Brodzik, K.; Nassour, I.; Yopp, A.; Singal, A.G.; Zhu, H.; Yu, H. Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; De Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef]
- Sieben, C.J.; Jeganathan, K.B.; Nelson, G.G.; Sturmlechner, I.; Zhang, C.; van Deursen, W.H.; Bakker, B.; Foijer, F.; Li, H.; Baker, D.J.; et al. BubR1 allelic effects drive phenotypic heterogeneity in mosaic-variegated aneuploidy progeria syndrome. J. Clin. Investig. 2020, 130, 171–188. [Google Scholar] [CrossRef]
- Bergeron, J.J.; Di Guglielmo, G.M.; Dahan, S.; Dominguez, M.; Posner, B.I. Spatial and temporal regulation of receptor tyrosine kinase activation and intracellular signal transduction. Annu. Rev. Biochem. 2016, 85, 573–597. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Villasenor, R.; Nonaka, H.; Del Conte-Zerial, P.; Kalaidzidis, Y.; Zerial, M. Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. eLife 2015, 4, e06156. [Google Scholar] [CrossRef]
- Jin, M.; Saucan, L.; Farquhar, M.G.; Palade, G.E. Rab1a and multiple other Rab proteins are associated with the transcytotic pathway in rat liver. J. Biol. Chem. 1996, 271, 30105–30113. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Škrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31, S262–S268. [Google Scholar] [CrossRef]
- Ader, M.; Stefanovski, D.; Kim, S.P.; Richey, J.M.; Ionut, V.; Catalano, K.J.; Hücking, K.; Ellmerer, M.; Van Citters, G.; Hsu, I.R.; et al. Hepatic insulin clearance is the primary determinant of insulin sensitivity in the normal dog. Obesity (Silver Spring) 2013, 22, 1238–1245. [Google Scholar] [CrossRef]
- Najjar, S.M.; Perdomo, G. Hepatic insulin clearance: Mechanism and physiology. Physiology (Bethesda) 2019, 34, 198–215. [Google Scholar] [CrossRef]
- Pawson, C.T.; Scott, J.D. Signal integration through blending, bolstering and bifurcating of intracellular information. Nat. Struct. Mol. Biol. 2010, 17, 653–658. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome structure and assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Filteau, M.; Vignaud, H.; Rochette, S.; Diss, G.; Chrétien, A.-È.; Berger, C.M.; Landry, C.R. Multi-scale perturbations of protein interactomes reveal their mechanisms of regulation, robustness and insights into genotype–phenotype maps. Brief. Funct. Genom. 2015, 15, 130–137. [Google Scholar] [CrossRef]
- Ge, H.; Liu, Z.; Church, G.M.; Vidal, M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 2001, 29, 482–486. [Google Scholar] [CrossRef]
- Simonis, N.; Gonze, D.; Orsi, C.; Van Helden, J.; Wodak, S.J. Modularity of the transcriptional response of protein complexes in yeast. J. Mol. Biol. 2006, 363, 589–610. [Google Scholar] [CrossRef]
- Gagnon-Arsenault, I.; Blanchet, F.-C.M.; Rochette, S.; Diss, G.; Dubé, A.K.; Landry, C.R. Transcriptional divergence plays a role in the rewiring of protein interaction networks after gene duplication. J. Proteom. 2013, 81, 112–125. [Google Scholar] [CrossRef]
- Hamdi, A.; Colas, P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol. Sci. 2012, 33, 109–118. [Google Scholar] [CrossRef]
- Zhong, Q.; Simonis, N.; Li, Q.; Charloteaux, B.; Heuze, F.; Klitgord, N.; Tam, S.; Yu, H.; Venkatesan, K.; Mou, D.; et al. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 2009, 5, 321. [Google Scholar] [CrossRef]
- Sahni, N.; Yi, S.; Taipale, M.; Bass, J.I.F.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef]
- Melamed, D.; Young, D.L.; Miller, C.R.; Fields, S. Combining natural sequence variation with high throughput mutational data to reveal protein interaction sites. PLoS Genet. 2015, 11, e1004918. [Google Scholar] [CrossRef]
- Starita, L.M.; Young, D.L.; Islam, M.; Kitzman, J.O.; Gullingsrud, J.; Hause, R.J.; Fowler, D.M.; Parvin, J.D.; Shendure, J.; Fields, S. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics 2015, 200, 413–422. [Google Scholar] [CrossRef]
- Sharma, A.; Menche, J.; Huang, C.C.; Ort, T.; Zhou, X.; Kitsak, M.; Sahni, N.; Thibault, D.; Voung, L.; Guo, F.; et al. A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma. Hum. Mol. Genet. 2015, 24, 3005–3020. [Google Scholar] [CrossRef]
- Boutchueng-Djidjou, M.; Faure, R.L. Network medicine-travelling with the insulin receptor: Encounter of the second type. EClinicalMedicine 2019, 13, 14–20. [Google Scholar] [CrossRef]
- Fernández-Tajes, J.; Gaulton, K.J.; Van De Bunt, M.; Torres, J.; Thurner, M.; Mahajan, A.; Gloyn, A.L.; Lage, K.; McCarthy, M.I. Developing a network view of type 2 diabetes risk pathways through integration of genetic, genomic and functional data. Genome Med. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Barrière, C.; Santamaría, D.; Cerqueira, A.; Galán, J.; Martín, A.; Ortega, S.; Malumbres, M.; Dubus, P.; Barbacid, M. Mice thrive without Cdk4 and Cdk2. Mol. Oncol. 2007, 1, 72–83. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, J.H.; Merrins, M.J.; Gavrilova, O.; Bisteau, X.; Kaldis, P.; Satin, L.S.; Rane, S.G. Loss of cyclin-dependent kinase 2 in the pancreas links primary beta-cell dysfunction to progressive depletion of beta-cell mass and diabetes. J. Biol. Chem. 2017, 292, 3841–3853. [Google Scholar] [CrossRef]
- Gaulin, J.F.; Fiset, A.; Fortier, S.; Faure, R.L. Characterization of Cdk2-cyclin E complexes in plasma membrane and endosomes of liver parenchyma. Insulin-dependent regulation. J. Biol. Chem. 2000, 275, 16658–16665. [Google Scholar] [CrossRef]
- Jongsma, M.L.; Berlin, I.; Neefjes, J. On the move: Organelle dynamics during mitosis. Trends Cell Biol. 2015, 25, 112–124. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef]
- Blake, J.A.; Eppig, J.T.; Kadin, J.A.; Richardson, J.E.; Smith, C.L.; Bult, C.J.; the Mouse Genome Database Group. Mouse genome database (MGD)-2017: Community knowledge resource for the laboratory mouse. Nucleic Acids Res. 2017, 45, D723–D729. [Google Scholar] [CrossRef] [PubMed]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Véronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; André, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228–237. [Google Scholar] [CrossRef]
- Boutchueng-Djidjou, M.; Collard-Simard, G.; Fortier, S.; Hébert, S.S.; Kelly, I.; Landry, C.R.; Faure, R.L. The last enzyme of the de novo purine synthesis pathway 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase (ATIC) plays a central role in insulin signaling and the golgi/endosomes protein network. Mol. Cell. Proteom. 2015, 14, 1079–1092. [Google Scholar] [CrossRef]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Berghe, G.V.D.; Vincent, M.-F. AICA-ribosiduria: A novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef]
- Ramond, F.; Rio, M.; Héron, B.; Imbard, A.; Marie, S.; Billiemaz, K.; Denommé-Pichon, A.; Kuentz, P.; Ceballos, I.; Piraud, M.; et al. AICA-ribosiduria due to ATIC deficiency: Delineation of the phenotype with three novel cases, and long-term update on the first case. J. Inherit. Metab. Dis. 2020, 43, 1254–1264. [Google Scholar] [CrossRef]
- Ikeda, M.; Kanao, Y.; Yamanaka, M.; Sakuraba, H.; Mizutani, Y.; Igarashi, Y.; Kihara, A. Characterization of four mammalian 3-hydroxyacyl-CoA dehydratases involved in very long-chain fatty acid synthesis. FEBS Lett. 2008, 582, 2435–2440. [Google Scholar] [CrossRef]
- David, D.; Sundarababu, S.; Gerst, J.E. Involvement of long chain fatty acid elongation in the trafficking of secretory vesicles in yeast. J. Cell Biol. 1998, 143, 1167–1182. [Google Scholar] [CrossRef]
- Obara, K.; Kojima, R.; Kihara, A. Effects on vesicular transport pathways at the late endosome in cells with limited very long-chain fatty acids. J. Lipid Res. 2013, 54, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Sawai, M.; Uchida, Y.; Ohno, Y.; Miyamoto, M.; Nishioka, C.; Itohara, S.; Sassa, T.; Kihara, A. The 3-hydroxyacyl-CoA dehydratases HACD1 and HACD2 exhibit functional redundancy and are active in a wide range of fatty acid elongation pathways. J. Biol. Chem. 2017, 292, 15538–15551. [Google Scholar] [CrossRef]
- Hirao, J.; Tojo, A.; Hatakeyama, S.; Satonaka, H.; Ishimitsu, T. V-ATPase blockade reduces renal gluconeogenesis and improves insulin secretion in type 2 diabetic rats. Hypertens. Res. 2020, 43, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P.; Hayles, J. The cell in an era of systems biology. Cell 2011, 144, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Wray, N.R.; Wijmenga, C.; Sullivan, P.F.; Yang, J.; Visscher, P.M. Common disease is more complex than implied by the core gene omnigenic model. Cell 2018, 173, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.I.; Pritchard, J.K. Trans effects on gene expression can drive omnigenic inheritance. Cell 2019, 177, 1022–1034.e6. [Google Scholar] [CrossRef]
- Chateigner, A.; Lesage-Descauses, M.-C.; Rogier, O.; Jorge, V.; Leplé, J.-C.; Brunaud, V.; Roux, C.P.-L.; Soubigou-Taconnat, L.; Martin-Magniette, M.-L.; Sanchez, L.; et al. Gene expression predictions and networks in natural populations supports the omnigenic theory. BMC Genom. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Maron, B.A.; Altucci, L.; Balligand, J.L.; Baumbach, J.; Ferdinandy, P.; Filetti, S.; Parini, P.; Petrillo, E.; Silverman, E.K.; Barabasi, A.L.; et al. A global network for network medicine. npj Syst. Biol. Appl. 2020, 6, 1–3. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leroux, M.; Boutchueng-Djidjou, M.; Faure, R. Insulin’s Discovery: New Insights on Its Hundredth Birthday: From Insulin Action and Clearance to Sweet Networks. Int. J. Mol. Sci. 2021, 22, 1030. https://doi.org/10.3390/ijms22031030
Leroux M, Boutchueng-Djidjou M, Faure R. Insulin’s Discovery: New Insights on Its Hundredth Birthday: From Insulin Action and Clearance to Sweet Networks. International Journal of Molecular Sciences. 2021; 22(3):1030. https://doi.org/10.3390/ijms22031030
Chicago/Turabian StyleLeroux, Melanie, Martial Boutchueng-Djidjou, and Robert Faure. 2021. "Insulin’s Discovery: New Insights on Its Hundredth Birthday: From Insulin Action and Clearance to Sweet Networks" International Journal of Molecular Sciences 22, no. 3: 1030. https://doi.org/10.3390/ijms22031030
APA StyleLeroux, M., Boutchueng-Djidjou, M., & Faure, R. (2021). Insulin’s Discovery: New Insights on Its Hundredth Birthday: From Insulin Action and Clearance to Sweet Networks. International Journal of Molecular Sciences, 22(3), 1030. https://doi.org/10.3390/ijms22031030