
Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System




Abstract
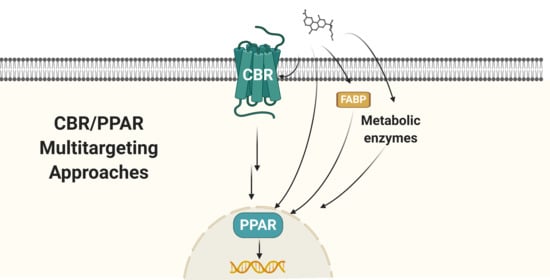
1. Introduction
2. CB1R–PPAR Modulation
2.1. CB1R–PPARα
2.2. CB1R-PPARγ
3. CB2R–PPAR Modulation
3.1. CB2R–PPARγ
3.2. CB2R–PPARα
4. FAAH–PPAR Modulation
5. Other CBR–PPAR Modulatory Profiles
5.1. CBR–PPARγ
5.2. CBR–PPARα
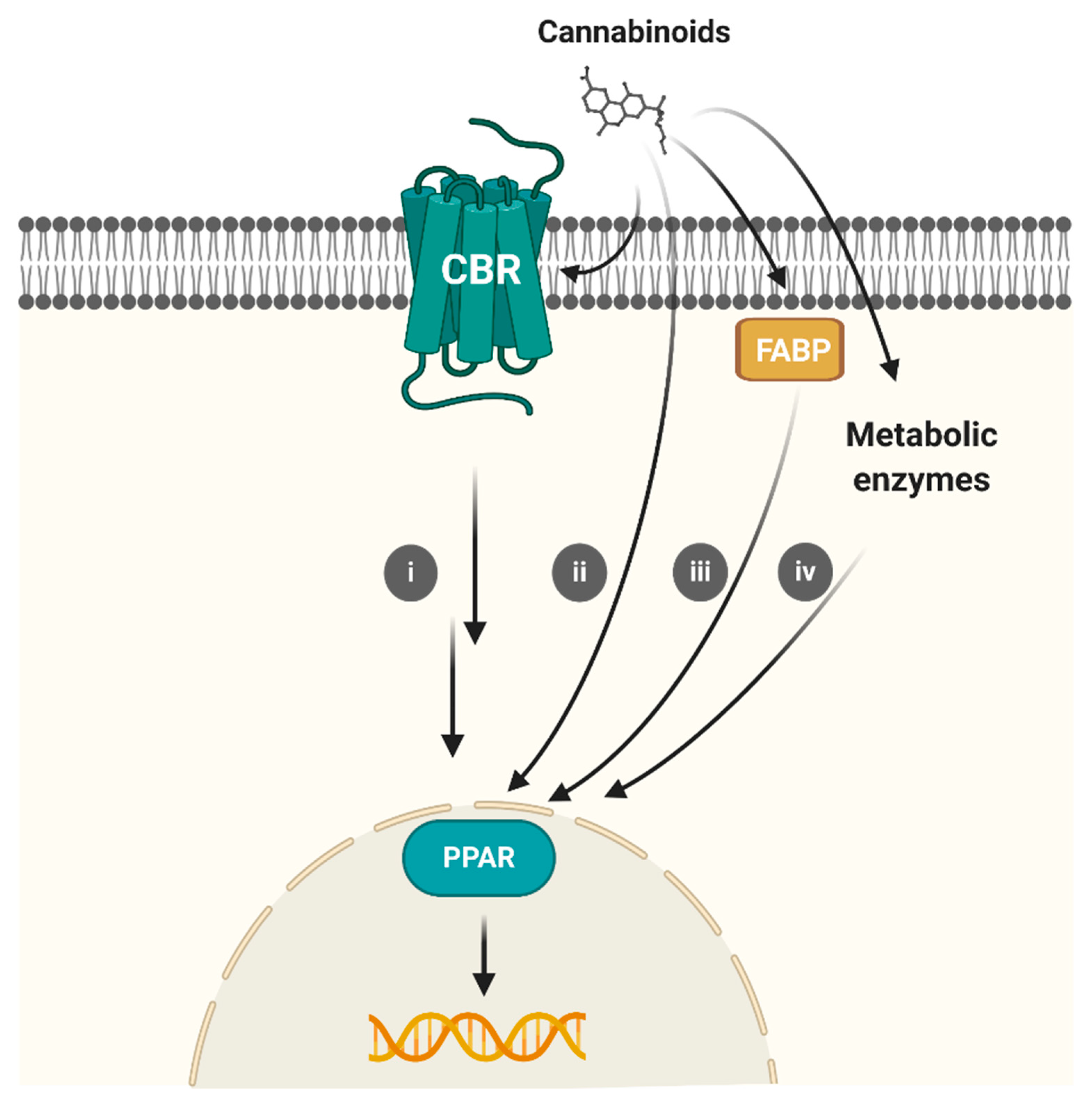
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 15d-PGJ2-G | 15-Desoxy-Δ12,14-prostaglandin-J2-glycerol ester |
| 2-AG | 2-Arachinoylglycerol |
| 5-HT1a | Serotonin 1A receptor |
| ABHD | α/β Hydrolase domain |
| AEA | N-Arachidonoyl-ethanolamine |
| AJA | Ajulemic acid |
| BBB | Blood brain barrier |
| BCP | β-Caryophyllene |
| CB1R | Cannabinoid receptor type 1 |
| CB2R | Cannabinoid receptor type 2 |
| CBC | Cannabichromene |
| CBCQ | Cannabichromenquinone |
| CBD | Cannabidiol |
| CBDA | Cannabidiolic acid |
| CBDQ | Cannabidiol hydroxyquinone |
| CBG | Cannabigerol |
| CBGA | Cannabigerolic acid |
| CBGQ | Cannabigeroquinone |
| CBM | Cannabimovone |
| CBN | Cannabinol |
| CBNQ | Cannabinolquinone |
| CBR | Cannabinoid receptor |
| Cryo-EM | Cryo electron microscopy |
| DGL | Diacylglycerol lipase |
| DIO | Diet-induced obese |
| ECS | Endocannabinoid System |
| EMI | Mangifera indica |
| EMT | Endocannabinoid membrane transporter |
| FA2H | Fatty acid 2-hydroxylase |
| FAAH | Fatty acid amide hydrolase |
| FABP | Fatty acid binding protein |
| GlyR | Glycine receptor |
| GPCR | G-Protein coupled receptor |
| LBD | Ligand binding domain |
| MAGL | Monoacylglycerol lipase |
| MD | Molecular dynamic |
| MHK | 4′-O-Methylhonokiol |
| NAAA | N-Acylethanolamine acid amidase |
| NAFLD | Non-alcoholic fatty liver disease |
| NAPE-PLD | N-Acyl phosphatidylethanolamine-specific phospholipase D |
| OEA | Oleoylethanolamide |
| OlGly | Oleoyl glycine |
| OLHHA | Oleic acid–dihydroxyamphetamine |
| PEA | Palmitoylethanolamide |
| PPAR | Peroxisome proliferator-activated receptors |
| PPRE | Peroxisome proliferator response element |
| THC | Δ9-Tetrahydrocannabinol |
| THCA | Δ9-Tetrahydrocannabinolic acid |
| TRP | Transient receptor potential |
| TRPM | Transient receptor potential melastatin |
| TZD | Thiazolidinedione |
| T2DM | Type 2 diabetes mellitus |
References
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Barlocco, D. Designed multiple ligands: Basic research vs. clinical outcomes. Curr. Med. Chem. 2012, 19, 3353–3387. [Google Scholar] [CrossRef]
- Naveja, J.J.; González, F.I.S.; Cruz, N.S.; Franco, J.L.M. Cheminformatics Approaches to Study Drug Polypharmacology. In Methods in Pharmacology and Toxicology; Springer: New York, NY, USA, 2018; pp. 3–25. [Google Scholar]
- Pertwee, R.G. (Ed.) Handbook of Cannabis; Oxford University Press: New York, NY, USA, 2014; ISBN 978-0-19-966268-5. [Google Scholar]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Devane, W.; Hanus, L.; Breuer, A.; Pertwee, R.; Stevenson, L.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shabat, S.B.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Simcocks, A.C.; Jenkin, K.A.; O’Keefe, L.; Samuel, C.S.; Mathai, M.L.; McAinch, A.J.; Hryciw, D.H. Atypical cannabinoid ligands O-1602 and O-1918 administered chronically in diet-induced obesity. Endocr. Connect. 2019, 8, 203–216. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Shaar, M.A. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Mechoulam, R.; Hanus, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef]
- di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Jagerovic, N. Novel approaches and current challenges with targeting the endocannabinoid system. Expert Opin. Drug Discov. 2020, 15, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Johnston, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Krishna Kumar, K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef]
- Xing, C.; Zhuang, Y.; Xing, C.; Zhuang, Y.; Xu, T.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-G i Signaling Complex. Cell 2020, 180, 645–654.e131. [Google Scholar] [CrossRef]
- Li, X.; Shen, L.; Hua, T.; Liu, Z.J. Structural and Functional Insights into Cannabinoid Receptors. Trends Pharmacol. Sci. 2020, 41, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Nomura, D.K.; Vann, R.E.; Walentiny, D.M.; Booker, L.; Jin, X.; Burston, J.J.; Sim-selley, L.J.; Lichtman, A.H.; Wiley, J.L.; et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20270–20275. [Google Scholar] [CrossRef] [PubMed]
- Abood, M.; Alexander, S.; Barth, F.; Bonner, T.; Bradshaw, H.; Cabral, G.; Casellas, P.; Cravatt, B.; Devane, W.; di Marzo, V.; et al. Cannabinoid Receptors (Version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide to Pharmacology CITE. 2019. Available online: https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=13 (accessed on 10 November 2020).
- Morales, P.; Reggio, P.H. An Update on Non-CB1, Non-CB2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Therapeutic Potential of Nonpsychoactive Cannabinoids by Targeting at Glycine Receptors. In Cannabinoids in Health and Disease; InTech: London, UK, 2016; p. 13. [Google Scholar]
- Xiong, W.; Cheng, K.; Cui, T.; Godlewski, G.; Rice, K.C.; Xu, Y.; Zhang, L. Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nat. Chem. Biol. 2011, 7, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Fernández-Dueñas, V.; López-Cano, M.; Taura, J.; Watanabe, M.; Ferrer, I.; Luján, R.; Ciruela, F. Adenosine A2A-Cannabinoid CB1 Receptor Heteromers in the Hippocampus: Cannabidiol Blunts Δ9-Tetrahydrocannabinol-Induced Cognitive Impairment. Mol. Neurobiol. 2019, 56, 5382–5391. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E.; Kendall, D.A. Cannabinoid activation of peroxisome proliferator-activated receptors: Potential for modulation of inflammatory disease. Immunobiology 2010, 215, 611–616. [Google Scholar] [CrossRef]
- Bagher, A.M.; Laprairie, R.B.; Kelly, M.E.M.; Wright, E.M.D. Antagonism of Dopamine Receptor 2 Long Affects Cannabinoid Receptor 1 Signaling in a Cell Culture Model of Striatal Medium Spiny Projection Neurons. Mol. Pharmacol. 2016, 89, 652–666. [Google Scholar] [CrossRef]
- Sierra, S.; Gupta, A.; Gomes, I.; Fowkes, M.; Ram, A.; Bobeck, E.N.; Devi, L.A. Targeting Cannabinoid 1 and Delta Opioid Receptor Heteromers Alleviates Chemotherapy-Induced Neuropathic Pain. ACS Pharmacol. Transl. Sci. 2019, 2, 219–229. [Google Scholar] [CrossRef]
- Franco, R.; Villa, M.; Morales, P.; Resina, I.R.; Rodríguez, A.G.; Jiménez, J.; Jagerovic, N.; Orgado, J.M.; Navarro, G. Increased expression of cannabinoid CB2 and serotonin 5-HT1A heteroreceptor complexes in a model of newborn hypoxic-ischemic brain damage. Neuropharmacology 2019, 152, 58–66. [Google Scholar] [CrossRef]
- Yousefnia, S.; Momenzadeh, S.; Forootan, F.S.; Ghaedi, K.; Esfahani, M.H.N. The influence of peroxisome proliferator-activated receptor γ (PPARγ) ligands on cancer cell tumorigenicity. Gene 2018, 649, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Decara, J.; Rivera, P.; Gambero, A.J.L.; Serrano, A.; Pavón, F.J.; Baixeras, E.; de Fonseca, F.R.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef]
- Pistis, M.; O’Sullivan, S.E. The Role of Nuclear Hormone Receptors in Cannabinoid Function, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 80, ISBN 1054-3589. [Google Scholar]
- Sun, Y.; Alexander, S.P.H.; Garle, M.J.; Gibson, C.L.; Hewitt, K.; Murphy, S.P.; Kendall, D.A.; Bennett, A.J. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br. J. Pharmacol. 2007, 152, 734–743. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoid activation of peroxisome proliferator-activated receptors: An update and review of the physiological relevance. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2013, 2, 17–25. [Google Scholar] [CrossRef]
- Yadav, M.R.; Murumkar, P.R. Advances in patented CB1 receptor antagonists for obesity. Pharm. Pat. Anal. 2018, 7, 169–173. [Google Scholar] [CrossRef]
- Murphy, T.; Foll, B.L. Targeting the endocannabinoid CB1 receptor to treat body weight disorders: A preclinical and clinical review of the therapeutic potential of past and present CB1 drugs. Biomolecules 2020, 10, 855. [Google Scholar] [CrossRef]
- Jagerovic, N.; Fernandez-fernandez, C.; Goya, P. CB1 Cannabinoid Antagonists: Structure-Activity Relationships and Potential Therapeutic Applications. Curr. Top. Med. Chem. 2008, 8, 205–230. [Google Scholar] [CrossRef]
- Stienstra, R.; Duval, C.; Müller, M.; Kersten, S. PPARs, obesity, and inflammation. PPAR Res. 2007, 2007, 95974. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M. The role of PPARα in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARα actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Almeida, B.; Joglar, J.; Rojas, M.J.L.; Decara, J.M.; Silva, F.J.B.; González, M.M.; Fitó, M.; Cuevas, M.R.; Farré, M.; Covas, M.I.; et al. Synthesis of fatty acid amides of catechol metabolites that exhibit antiobesity properties. ChemMedChem 2010, 5, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- de la Torre Fornell, R.; Albadalejo, M.F.; Planells, M.I.C.; Colomert, M.F.; Cotrim, B.A.; de Fonseca, F.R.; del Olmo, J.M.D.; González, M.M.; Cuevas, M.R.; Amargo, J.J.; et al. Fatty Acid Amide Derivatives with Amphetamines for the Treatment of Eating. Disorders. Patent Publication Number WO 2011/076966, 30 June 2011. [Google Scholar]
- Decara, J.M.; Pavón, F.J.; Suárez, J.; Cuevas, M.R.; Baixeras, E.; Vázquez, M.; Rivera, P.; Gavito, A.L.; Almeida, B.; Joglar, J.; et al. Treatment with a novel oleic-acid-dihydroxyamphetamine conjugation ameliorates non-alcoholic fatty liver disease in obese Zucker rats. DMM Dis. Model. Mech. 2015, 8, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Alen, F.; Decara, J.; Brunori, G.; You, Z.B.; Bühler, K.M.; Moreno, J.A.L.; Cippitelli, A.; Pavon, F.J.; Suárez, J.; Gardner, E.L.; et al. PPARα/CB1 receptor dual ligands as a novel therapy for alcohol use disorder: Evaluation of a novel oleic acid conjugate in preclinical rat models. Biochem. Pharmacol. 2018, 157, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.P.; Fresno, N.; González, M.M.; Elguero, J.; Decara, J.; Girón, R.; Rodríguez-Álvarez, A.; Martín, M.I.; Rodríguez De Fonseca, F.; Goya, P. Discovery of potent dual PPARα agonists/CB1 ligands. ACS Med. Chem. Lett. 2011, 2, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Azar, S.; Udi, S.; Drori, A.; Hadar, R.; Nemerovski, A.; Vemuri, K.V.; Miller, M.; Rofe, D.S.; Arad, Y.; Wahnon, D.G.; et al. Reversal of Diet-induced Hepatic Steatosis by Peripheral CB1 Receptor Blockade in Mice is p53/miRNA-22/SIRT1/PPARα Dependent. Mol. Metab. 2020, 42, 101087. [Google Scholar] [CrossRef]
- Serrano, A.; del Arco, I.; Pavón, F.J.; Macías, M.; Valero, V.P.; de Fonseca, F.R. The cannabinoid CB1 receptor antagonist SR141716A (Rimonabant) enhances the metabolic benefits of long-term treatment with oleoylethanolamide in Zucker rats. Neuropharmacology 2008, 54, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Morell, C.; Henche, N.R.; Laviada, I.D. Involvement of PPARγ in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis. 2013, 4, e618. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Gaetani, S.; Oveisi, F.; Verme, J.L.; Serrano, A.; de Fonseca, F.R.; Rosengarth, A.; Luecke, H.; di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Panikashvili, D.; Mechoulam, R.; Beni, S.M.; Alexandrovich, A.; Shohami, E. CB1 cannabinoid receptors are involved in neuroprotection via NF-κB inhibition. J. Cereb. Blood Flow Metab. 2005, 25, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Van der Stelt, M.; di Marzo, V. Cannabinoid receptors and their role in neuroprotection. Neuro. Mol. Med. 2005, 7, 37–50. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed]
- Bordet, R.; Gelé, P.; Duriez, P.; Fruchart, J.C. PPARs: A new target for neuroprotection. J. Neurol. Neurosurg. Psychiatry 2006, 77, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Citraro, R.; Russo, E.; Scicchitano, F.; van Rijn, C.M.; Cosco, D.; Avagliano, C.; Russo, R.; D’Agostino, G.; Petrosino, S.; Guida, F.; et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology 2013, 69, 115–126. [Google Scholar] [CrossRef]
- Lambert, D.M.; Di Marzo, V. The palmitoylethanolamide and oleamide enigmas: Are these two fatty acid amides cannabimimetic? Curr. Med. Chem. 1999, 6, 757–773. [Google Scholar]
- Ho, W.S.V.; Barrett, D.A.; Randall, M.D. “Entourage” effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br. J. Pharmacol. 2008, 155, 837–846. [Google Scholar] [CrossRef]
- Jonsson, K.O.; Vandevoorde, S.; Lambert, D.M.; Tiger, G.; Fowler, C.J. Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoid anandamide. Br. J. Pharmacol. 2001, 133, 1263–1275. [Google Scholar] [CrossRef]
- Alsalem, M.; Haddad, M.; Aldossary, S.A.; Kalbouneh, H.; Altarifi, A.; Jaffal, S.M.; Abbas, M.A.; Aldaoud, N.; El-Salem, K. Role of cannabinoid receptor 1 and the peroxisome proliferator-activated receptor α in mediating anti-nociceptive effects of synthetic cannabinoids and a cannabinoid-like compound. Inflammopharmacology 2019, 27, 1131–1142. [Google Scholar] [CrossRef]
- Russo, R.; Verme, J.L.; Rana, G.L.; D’Agostino, G.; Sasso, O.; Calignano, A.; Piomelli, D. Synergistic antinociception by the cannabinoid receptor agonist anandamide and the PPAR-α receptor agonist GW7647. Eur. J. Pharmacol. 2007, 566, 117–119. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on peroxisome proliferator-activated receptor (PPAR) activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the endocannabinoid system as a potential anticancer strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Dariš, B.; Verboten, M.T.; Knez, Ž.; Ferk, P. Cannabinoids in cancer treatment: Therapeutic potential and legislation. Bosn. J. Basic Med. Sci. 2019, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The endocannabinoid system: A target for cancer treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef]
- Morales, P.; Vara, D.; Cañas, M.G.; Zúñiga, M.C.; Azar, C.O.; Goya, P.; Ruiz, J.F.; Laviada, I.D.; Jagerovic, N. Synthetic cannabinoid quinones: Preparation, in vitro antiproliferative effects and in vivo prostate antitumor activity. Eur. J. Med. Chem. 2013, 70, 111–119. [Google Scholar] [CrossRef]
- Seltzer, E.S.; Watters, A.K.; Mackenzie, D.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a promising anti-cancer drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Aviello, G.; Romano, B.; Borrelli, F.; Capasso, R.; Gallo, L.; Piscitelli, F.; Di Marzo, V.; Izzo, A.A. Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J. Mol. Med. 2012, 90, 925–934. [Google Scholar] [CrossRef]
- O’Sullivan, S.E.; Sun, Y.; Bennett, A.J.; Randall, M.D.; Kendall, D.A. Time-dependent vascular actions of cannabidiol in the rat aorta. Eur. J. Pharmacol. 2009, 612, 61–68. [Google Scholar] [CrossRef]
- McPartland, J.M.; Glass, M.; Pertwee, R.G. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: Interspecies differences. Br. J. Pharmacol. 2007, 152, 583–593. [Google Scholar] [CrossRef]
- Ramer, R.; Heinemann, K.; Merkord, J.; Rohde, H.; Salamon, A.; Linnebacher, M.; Hinz, B. COX-2 and PPAR-γ confer cannabidiol-induced apoptosis of human lung cancer cells. Mol. Cancer Ther. 2013, 12, 69–82. [Google Scholar] [CrossRef]
- Brito, L.F.; Gontijo, D.C.; Toledo, R.C.L.; Barcelos, R.M.; de Oliveira, A.B.; Brandão, G.C.; de Sousa, L.P.; Ribeiro, S.M.R.; Leite, J.P.V.; Fietto, L.G.; et al. Mangifera indica leaves extract and mangiferin modulate CB1 and PPARγ receptors and others markers associated with obesity. J. Funct. Foods 2019, 56, 74–83. [Google Scholar] [CrossRef]
- Fahmi, H.; Pelletier, J.M.; Pelletier, J.-P.; Kapoor, M. Peroxisome proliferator-activated receptor gamma in osteoarthritis. Mod. Rheumatol. 2011, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marder, W.; Khalatbari, S.; Myles, J.D.; Hench, R.; Lustig, S.; Yalavarthi, S.; Parameswaran, A.; Brook, R.D.; Kaplan, M.J. The peroxisome proliferator activated receptor-γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J. Am. Heart Assoc. 2013, 2, e000441. [Google Scholar] [CrossRef]
- Lowin, T.; Straub, R.H. Cannabinoid-based drugs targeting CB1 and TRPV1, the sympathetic nervous system, and arthritis. Arthritis Res. Ther. 2015, 17, 226. [Google Scholar] [CrossRef] [PubMed]
- Palomares, B.; Rodriguez, M.G.; Consuegra, C.G.; Cañas, M.G.; Saen-oon, S.; Soliva, R.; Collado, J.A.; Ruiz, J.F.; Morello, G.; Calzado, M.A.; et al. Δ9-Tetrahydrocannabinolic acid alliviates collagen-induced arthritis: Role of PPARγ and CB1 receptors. Br. J. Pharmacol. 2020, 177, 4034–4054. [Google Scholar] [CrossRef]
- Nadal, X.; del Río, C.; Casano, S.; Palomares, B.; Vera, C.F.; Navarrete, C.; Carnerero, C.S.; Cantarero, I.; Bellido, M.L.; Meyer, S.; et al. Tetrahydrocannabinolic acid is a potent PPARγ agonist with neuroprotective activity. Br. J. Pharmacol. 2017, 174, 4263–4276. [Google Scholar] [CrossRef]
- Stebulis, J.A.; Johnson, D.R.; Rossetti, R.G.; Burstein, S.H.; Zurier, R.B. Ajulemic acid, a synthetic cannabinoid acid, induces an antiinflammatory profile of eicosanoids in human synovial cells. Life Sci. 2008, 83, 666–670. [Google Scholar] [CrossRef]
- Irrera, N.; D’ascola, A.; Pallio, G.; Bitto, A.; Mazzon, E.; Mannino, F.; Squadrito, V.; Arcoraci, V.; Minutoli, L.; Campo, G.M.; et al. β-Caryophyllene Mitigates Collagen Antibody Induced Arthritis (CAIA) in Mice Through a Cross-Talk between CB2 and PPAR-γ Receptors. Biomolecules 2019, 9, 326. [Google Scholar] [CrossRef]
- Gonzalez, E.G.; Selvi, E.; Balistreri, E.; Akhmetshina, A.; Palumbo, K.; Lorenzini, S.; Lazzerini, P.E.; Montilli, C.; Capecchi, P.L.; Lucattelli, M.; et al. Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1545–1551. [Google Scholar] [CrossRef]
- del Rio, C.; Cantarero, I.; Palomares, B.; Cañas, M.G.; Ruiz, J.F.; Pavicic, C.; Martín, A.G.; Bellido, M.L.; Castro, R.O.; Sánchez, C.P.; et al. VCE-004.3, a cannabidiol aminoquinone derivative, prevents bleomycin-induced skin fibrosis and inflammation through PPARγ- and CB2 receptor-dependent pathways. Br. J. Pharmacol. 2018, 175, 3813–3831. [Google Scholar] [CrossRef]
- del Río, C.; Navarrete, C.; Collado, J.A.; Bellido, M.L.; Cañas, M.G.; Pazos, M.R.; Ruiz, J.F.; Pollastro, F.; Appendino, G.; Calzado, M.A.; et al. The cannabinoid quinol VCE-004.8 alleviates bleomycin-induced scleroderma and exerts potent antifibrotic effects through peroxisome proliferator-activated receptor-γ and CB2 pathways. Sci. Rep. 2016, 6, 21703. [Google Scholar] [CrossRef] [PubMed]
- Martín, A.G.; Rodríguez, M.G.; Navarrete, C.; del Río, C.; Bellido, M.L.; Appendino, G.; Calzado, M.A.; Muñoz, E. EHP-101, an oral formulation of the cannabidiol aminoquinone VCE-004.8, alleviates bleomycin-induced skin and lung fibrosis. Biochem. Pharmacol. 2018, 157, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, C.; Martin, A.G.; Rodríguez, M.G.; Mestre, L.; Feliú, A.; Guaza, C.; Calzado, M.A.; Muñoz, E. Effects of EHP-101 on inflammation and remyelination in murine models of Multiple sclerosis. Neurobiol. Dis. 2020, 143, 104994. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, C.; Salinas, F.C.; Palomares, B.; Mecha, M.; Jiménez, C.J.; Mestre, L.; Feliú, A.; Bellido, M.L.; Fiebich, B.L.; Appendino, G.; et al. Hypoxia mimetic activity of VCE-004.8, a cannabidiol quinone derivative: Implications for multiple sclerosis therapy. J. Neuroinflamm. 2018, 15, 64. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Choi, I.S.; Park, M.H.; Lee, Y.M.; Song, J.K.; Kim, Y.H.; Kim, K.H.; Hwang, D.Y.; Jeong, J.H.; Yun, Y.P.; et al. 4-O-Methylhonokiol attenuates memory impairment in presenilin 2 mutant mice through reduction of oxidative damage and inactivation of astrocytes and the ERK pathway. Free Radic. Biol. Med. 2011, 50, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dong, Z.; Liu, S. β-caryophyllene ameliorates the Alzheimer-like phenotype in APP/PS1 mice through CB2 receptor activation and the PPARγ pathway. Pharmacology 2014, 94, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Javed, H.; Azimullah, S.; Haque, M.E. β-Caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol. Cell. Biochem. 2016, 418, 59–70. [Google Scholar] [CrossRef]
- Mansouri, S.A.; Ojha, S.; Maamari, E.A.; Ameri, M.A.; Nurulain, S.M.; Bahi, A. The cannabinoid receptor 2 agonist, β-caryophyllene, reduced voluntary alcohol intake and attenuated ethanol-induced place preference and sensitivity in mice. Pharmacol. Biochem. Behav. 2014, 124, 260–268. [Google Scholar] [CrossRef]
- Galaj, E.; Bi, G.H.; Moore, A.; Chen, K.; He, Y.; Gardner, E.; Xi, Z.X. Beta-caryophyllene inhibits cocaine self-administration by activation of PPARα and PPARγ: Repurposing a FDA-approved food additive for cocaine use disorder. Neuropsychopharmacology 2020, 1–11. [Google Scholar] [CrossRef]
- Recht, L.D.; Salmonsen, R.; Rosetti, R.; Jang, T.; Pipia, G.; Kubiatowski, T.; Karim, P.; Ross, A.H.; Zurier, R.; Scott Litofsky, N.; et al. Antitumor effects of ajulemic acid (CT3), a synthetic non-psychoactive cannabinoid. Biochem. Pharmacol. 2001, 62, 755–763. [Google Scholar] [CrossRef]
- Hyun, S.; Kim, M.S.; Song, Y.S.; Bak, Y.; Ham, S.Y.; Lee, D.H.; Hong, J.; Yoon, D.Y. Peroxisome Proliferator-Activated Receptor-Gamma Agonist 4-O-Methylhonokiol Induces Apoptosis by Triggering the Intrinsic Apoptosis Pathway and Inhibiting the PI3K/Akt Survival Pathway in SiHa Human Cervical Cancer Cells. J. Microbiol. Biotechnol. 2015, 25, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; D’ascola, A.; Pallio, G.; Bitto, A.; Mannino, F.; Arcoraci, V.; Rottura, M.; Ieni, A.; Minutoli, L.; Metro, D.; et al. β-caryophyllene inhibits cell proliferation through a direct modulation of CB2 receptors in glioblastoma cells. Cancers 2020, 12, 1038. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhou, Y.; Wang, Y.; Xiao, S.; Liao, D.J.; Zhao, Q. PPARγ mediates the effects of WIN55,212-2, an synthetic cannabinoid, on the proliferation and apoptosis of the BEL-7402 hepatocarcinoma cells. Mol. Biol. Rep. 2013, 40, 6287–6293. [Google Scholar] [CrossRef]
- Palomares, B.; Pino, F.R.; Navarrete, C.; Velasco, I.; Garrido, M.A.S.; Jimenez, C.J.; Pavicic, C.; Vazquez, M.J.; Appendino, G.; Bellido, M.L.; et al. VCE-004.8, A Multitarget Cannabinoquinone, Attenuates Adipogenesis and Prevents Diet-Induced Obesity. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, J.; Jiang, X.; Wang, J.; Yan, X.; Zheng, Y.; Conklin, D.J.; Kim, K.S.; Kim, K.H.; Tan, Y.; et al. The magnolia bioactive constituent 4-O-methylhonokiol protects against high-fat diet-induced obesity and systemic insulin resistance in mice. Oxid. Med. Cell. Longev. 2014, 2014, 965954. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Hashiesh, H.M.; Meeran, M.F.N.; Sharma, C.; Sadek, B.; Kaabi, J.A.; Ojha, S.K. Therapeutic Potential of β-Caryophyllene: A Dietary Cannabinoid in Diabetes and Associated Complications. Nutrients 2020, 12, 2963. [Google Scholar] [CrossRef]
- Wu, K.; Xiu, Y.; Zhou, P.; Qiu, Y.; Li, Y. A New Use for an Old Drug: Carmofur Attenuates Lipopolysaccharide (LPS)-Induced Acute Lung Injury via Inhibition of FAAH and NAAA Activities. Front. Pharmacol. 2019, 10, 818. [Google Scholar] [CrossRef]
- Malamas, M.S.; Farah, S.I.; Lamani, M.; Pelekoudas, D.N.; Perry, N.T.; Rajarshi, G.; Miyabe, C.Y.; Chandrashekhar, H.; West, J.; Pavlopoulos, S.; et al. Design and synthesis of cyanamides as potent and selective N-acylethanolamine acid amidase inhibitors. Bioorganic Med. Chem. 2020, 28, 115195. [Google Scholar] [CrossRef]
- Rock, E.M.; Limebeer, C.L.; Ward, J.M.; Cohen, A.; Grove, K.; Niphakis, M.J.; Cravatt, B.F.; Parker, L.A. Interference with acute nausea and anticipatory nausea in rats by fatty acid amide hydrolase (FAAH) inhibition through a PPARα and CB1 receptor mechanism, respectively: A double dissociation. Psychopharmacology 2015, 232, 3841–3848. [Google Scholar] [CrossRef]
- Fotio, Y.; Palese, F.; Tipan, P.G.; Ahmed, F.; Piomelli, D. Inhibition of fatty acid amide hydrolase in the CNS prevents and reverses morphine tolerance in male and female mice. Br. J. Pharmacol. 2020, 177, 3024–3035. [Google Scholar] [CrossRef]
- Ayoub, S.M.; Smoum, R.; Farag, M.; Atwal, H.; Collins, S.A.; Rock, E.M.; Limebeer, C.L.; Piscitelli, F.; Iannotti, F.A.; Lichtman, A.H.; et al. Oleoyl alanine (HU595): A stable monomethylated oleoyl glycine interferes with acute naloxone precipitated morphine withdrawal in male rats. Psychopharmacology 2020, 237, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.M.; Ayoub, S.M.; Limebeer, C.L.; Gene, A.; Wills, K.L.; de Vuono, M.V.; Smoum, R.; Di Marzo, V.; Lichtman, A.H.; Mechoulam, R.; et al. Acute naloxone-precipitated morphine withdrawal elicits nausea-like somatic behaviors in rats in a manner suppressed by N-oleoylglycine. Psychopharmacology 2020, 237, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Donvito, G.; Piscitelli, F.; Muldoon, P.; Jackson, A.; Vitale, R.M.; D’Aniello, E.; Giordano, C.; Ignatowska-Jankowska, B.M.; Mustafa, M.A.; Guida, F.; et al. N-Oleoyl-glycine reduces nicotine reward and withdrawal in mice. Neuropharmacology 2019, 148, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Raymundi, A.M.; da Silva, T.R.; Zampronio, A.R.; Guimarães, F.S.; Bertoglio, L.J.; Stern, C.A.J. A time-dependent contribution of hippocampal CB1, CB2 and PPARγ receptors to cannabidiol-induced disruption of fear memory consolidation. Br. J. Pharmacol. 2020, 177, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; de Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 2012, 63, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.D.; Luna, J.P.; Navarrete, C.; del Río, C.; Cantarero, I.; Palomares, B.; Aguareles, J.; Ruiz, J.F.; Bellido, M.L.; Pollastro, F.; et al. VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Sci. Rep. 2016, 6, 29789. [Google Scholar] [CrossRef]
- Aguareles, J.; Luna, J.P.; Palomares, B.; Grañeras, R.B.; Navarrete, C.; Calvo, A.R.; Rincón, D.G.; Taboada, E.G.; Guzmán, M.; Muñoz, E.; et al. Oral administration of the cannabigerol derivative VCE-003.2 promotes subventricular zone neurogenesis and protects against mutant huntingtin-induced neurodegeneration. Transl. Neurodegener. 2019, 8, 9. [Google Scholar] [CrossRef]
- García, C.; Cañas, M.G.; Burgaz, S.; Palomares, B.; Gálvez, Y.G.; Garo, C.P.; Campo, S.; Hernández, J.F.; Pavicic, C.; Navarrete, C.; et al. Benefits of VCE-003.2, a cannabigerol quinone derivative, against inflammation-driven neuronal deterioration in experimental Parkinson’s disease: Possible involvement of different binding sites at the PPARγ receptor. J. Neuroinflamm. 2018, 15, 19. [Google Scholar] [CrossRef]
- Burgaz, S.; García, C.; Gómez-Cañas, M.; Muñoz, E.; Ruiz, J.F. Development of An Oral Treatment with the PPAR-γ-Acting Cannabinoid VCE-003.2 Against the Inflammation-Driven Neuronal Deterioration in Experimental P arkinson’s Disease. Molecules 2019, 24, 2702. [Google Scholar] [CrossRef] [PubMed]
- Granja, A.G.; Salinas, F.C.; Pagani, A.; Cañas, M.G.; Negri, R.; Navarrete, C.; Mecha, M.; Mestre, L.; Fiebich, L.; Cantarero, I.; et al. A Cannabigerol Quinone Alleviates Neuroinflammation in a Chronic Model of Multiple Sclerosis. J. Neuroimmune Pharmacol. 2012, 7, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Hind, W.H.; England, T.J.; O’Sullivan, S.E. Cannabidiol protects an in vitro model of the blood-brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT1A receptors. Br. J. Pharmacol. 2016, 173, 815–825. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E.; Tarling, E.J.; Bennett, A.J.; Kendall, D.A.; Randall, M.D. Novel time-dependent vascular actions of Δ9- tetrahydrocannabinol mediated by peroxisome proliferator-activated receptor gamma. Biochem. Biophys. Res. Commun. 2005, 337, 824–831. [Google Scholar] [CrossRef]
- Enayatfard, L.; Rostami, F.; Nasoohi, S.; Oryan, S.; Ahmadiani, A.; Dargahi, L. Dual role of PPAR-γ in induction and expression of behavioral sensitization to cannabinoid receptor agonist WIN55,212-2. Neurol. Mol. Med. 2013, 15, 523–535. [Google Scholar] [CrossRef]
- Suzuki, M.H.; Takeda, S.; Watanabe, K.; Takiguchi, M.; Aramaki, H. Δ 9-Tetrahydrocannabinol upregulates fatty acid 2-hydroxylase (FA2H) via PPARα induction: A possible evidence for the cancellation of PPARβ/δ-mediated inhibition of PPARα in MDA-MB-231cells. Arch. Biochem. Biophys. 2019, 662, 219–225. [Google Scholar] [CrossRef]
- Payandemehr, B.; Ebrahimi, A.; Gholizadeh, R.; Rahimian, R.; Varastehmoradi, B.; Gooshe, M.; Aghaei, H.N.; Mousavizadeh, K.; Dehpour, A.R. Involvement of PPAR receptors in the anticonvulsant effects of a cannabinoid agonist, WIN 55,212-2. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2015, 57, 140–145. [Google Scholar] [CrossRef]
- Couch, D.G.; Cook, H.; Ortori, C.; Barrett, D.; Lund, J.N.; O’Sullivan, S.E. Palmitoylethanolamide and Cannabidiol Prevent Inflammation-induced Hyperpermeability of the Human Gut In Vitro and In Vivo—A Randomized, Placebo-controlled, Double-blind Controlled Trial. Inflamm. Bowel Dis. 2019, 25, 1006–1018. [Google Scholar] [CrossRef]
- Rock, E.M.; Sullivan, M.T.; Pravato, S.; Pratt, M.; Limebeer, C.L.; Parker, L.A. Effect of combined doses of Δ9-tetrahydrocannabinol and cannabidiol or tetrahydrocannabinolic acid and cannabidiolic acid on acute nausea in male Sprague-Dawley rats. Psychopharmacology 2020, 237, 901–914. [Google Scholar] [CrossRef]
- Bai, J.; Ge, G.; Wang, Y.; Zhang, W.; Wang, Q.; Wang, W.; Guo, X.; Yu, B.; Xu, Y.; Yang, H.; et al. A selective CB2 agonist protects against the inflammatory response and joint destruction in collagen-induced arthritis mice. Biomed. Pharmacother. 2019, 116, 109025. [Google Scholar] [CrossRef]
- Fukuda, S.; Kohsaka, H.; Takayasu, A.; Yokoyama, W.; Miyabe, C.; Miyabe, Y.; Harigai, M.; Miyasaka, N.; Nanki, T. Cannabinoid receptor 2 as a potential therapeutic target in rheumatoid arthritis. BMC Musculoskelet. Disord. 2014, 15, 275. [Google Scholar] [CrossRef] [PubMed]
- Lowin, T.; Schneider, M.; Pongratz, G. Joints for joints: Cannabinoids in the treatment of rheumatoid arthritis. Curr. Opin. Rheumatol. 2019, 31, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hounoki, H.; Sugiyama, E.; Mohamed, S.G.K.; Shinoda, K.; Taki, H.; Abdel-Aziz, H.O.; Maruyama, M.; Kobayashi, M.; Miyahara, T. Activation of peroxisome proliferator-activated receptor γ inhibits TNF-α-mediated osteoclast differentiation in human peripheral monocytes in part via suppression of monocyte chemoattractant protein-1 expression. Bone 2008, 42, 765–774. [Google Scholar] [CrossRef]
- Ji, J.D.; Cheon, H.; Jun, J.B.; Choi, S.J.; Kim, Y.R.; Lee, Y.H.; Kim, T.H.; Chae, I.J.; Song, G.G.; Yoo, D.H.; et al. Effects of peroxisome proliferator-activated receptor-γ (PPAR-γ) on the expression of inflammatory cytokines and apoptosis induction in rheumatoid synovial fibroblasts and monocytes. J. Autoimmun. 2001, 17, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Burstein, S.H.; Zurier, R.B.; Chen, J.D. Activation and binding of peroxisome proliferator-activated receptor γ by synthetic cannabinoid ajulemic acid. Mol. Pharmacol. 2003, 63, 983–992. [Google Scholar] [CrossRef]
- Parker, J.; Atez, F.; Rossetti, R.G.; Skulas, A.; Patel, R.; Zurier, R.B. Suppression of human macrophage interleukin-6 by a nonpsychoactive cannabinoid acid. Rheumatol. Int. 2008, 28, 631–635. [Google Scholar] [CrossRef]
- Zurier, R.B.; Rossetti, R.G.; Lane, J.H.; Goldberg, J.M.; Hunter, S.A.; Burstein, S.H. Dimethylheptyl-THC-11 OIC acid: A nonpsychoactive antiinflammatory agent with a cannabinoid template structure. Arthritis Rheum. 1998, 41, 163–170. [Google Scholar] [CrossRef]
- Zurier, R.B.; Rossetti, R.G.; Burstein, S.H.; Bidinger, B. Suppression of human monocyte interleukin-1β production by ajulemic acid, a nonpsychoactive cannabinoid. Biochem. Pharmacol. 2003, 65, 649–655. [Google Scholar] [CrossRef]
- Burstein, S.H.; Audette, C.A.; Doyle, S.A.; Breuer, A.; Devane, W.A.; Colodner, S.; Mechoulam, R. Synthetic Nonpsychotropic Cannabinoids with Potent Antiinflammatory, Analgesic, and Leukocyte Antiadhesion Activities. J. Med. Chem. 1992, 35, 3135–3141. [Google Scholar] [CrossRef]
- Burstein, S. Ajulemic Acid (IP-751): Synthesis, Proof of Principle, Toxicity Studies, and Clinical Trials. AAPS J. 2005, 7, 143–148. [Google Scholar] [CrossRef]
- Karmakar, S.; Kay, J.; Gravallese, E.M. Bone Damage in rheumatoid arthritis: Mechanistic insights and approaches to prevention. Rheum. Dis. Clin. N. Am. 2010, 36, 385–404. [Google Scholar] [CrossRef] [PubMed]
- George, K.L.; Saltman, L.H.; Stein, G.S.; Lian, J.B.; Zurier, R.B. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J. Cell. Physiol. 2008, 214, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.A.; Fayoumi, H.M.E.; Mahmoud, M.F. Beta-caryophyllene alleviates diet-induced neurobehavioral changes in rats: The role of CB2 and PPAR-γ receptors. BioMed Pharmacother. 2019, 110, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jia, Y.; Lee, J.H.; Jun, H.J.; Lee, H.S.; Hwang, K.Y.; Lee, S.J. Trans-Caryophyllene is a natural agonistic ligand for peroxisome proliferator-activated receptor-α. Bioorganic Med. Chem. Lett. 2014, 24, 3168–3174. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, S.; Zhang, Q.; Yi, C.; He, J.; Ye, X.; Liu, M.; Lu, W. Celastrol is a novel selective agonist of cannabinoid receptor 2 with anti-inflammatory and anti-fibrotic activity in a mouse model of systemic sclerosis. Phytomedicine 2020, 67, 153160. [Google Scholar] [CrossRef]
- Servettaz, A.; Kavian, N.; Nicco, C.; Deveaux, V.; Chéreau, C.; Wang, A.; Zimmer, A.; Lotersztajn, S.; Weill, B.; Batteux, F. Targeting the cannabinoid pathway limits the development of fibrosis and autoimmunity in a mouse model of systemic sclerosis. Am. J. Pathol. 2010, 177, 187–196. [Google Scholar] [CrossRef]
- Gonzalez, E.G.; Selvi, E.; Balistreri, E.; Lorenzini, S.; Maggio, R.; Natale, M.R.; Capecchi, P.L.; Lazzerini, P.E.; Bardelli, M.; Laghi-Pasini, F.; et al. Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology 2009, 48, 1050–1056. [Google Scholar] [CrossRef]
- Dantas, A.T.; Pereira, M.C.; de Melo Rego, M.J.B.; da Rocha, L.F.; Pitta, I.D.R.; Marques, C.D.L.; Duarte, A.L.B.P.; Pitta, M.G.D.R. The Role of PPAR Gamma in Systemic Sclerosis. PPAR Res. 2015, 2015, 124624. [Google Scholar] [CrossRef]
- Lucattelli, M.; Fineschi, S.; Selvi, E.; Gonzalez, E.G.; Bartalesi, B.; de Cunto, G.; Lorenzini, S.; Galeazzi, M.; Lungarella, G. Ajulemic acid exerts potent anti-fibrotic effect during the fibrogenic phase of bleomycin lung. Respir. Res. 2016, 17, 49. [Google Scholar] [CrossRef]
- Spiera, R.; Hummers, L.; Chung, L.; Frech, T.M.; Domsic, R.; Hsu, V.; Furst, D.E.; Gordon, J.; Mayes, M.; Simms, R.; et al. Safety and Efficacy of Lenabasum in a Phase II, Randomized, Placebo-Controlled Trial in Adults with Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 1350–1360. [Google Scholar] [CrossRef]
- To Evaluate the Safety and Tolerability, Pharmacokinetics, Food-effect and Pharmacodynamics of EHP-101 in Healthy Volunteers. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03745001?term=EHP-101&draw=2&rank=2 (accessed on 9 November 2020).
- Alberti, T.B.; Barbosa, W.L.R.; Vieira, J.L.F.; Raposo, N.R.B.; Dutra, R.C. (−)-β-caryophyllene, a CB2 receptor-selective phytocannabinoid, suppresses motor paralysis and neuroinflammation in a murine model of multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 691. [Google Scholar] [CrossRef] [PubMed]
- Fontes, L.B.A.; Dias, D.; Dos, S.; Aarestrup, B.J.V.; Aarestrup, F.M.; da Silva Filho, A.A.; do Corrêa, J.O.A. β-Caryophyllene ameliorates the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. BioMed Pharmacother. 2017, 91, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Nick, R.S. Promising neuroprotective effects of β-caryophyllene against LPS-induced oligodendrocyte toxicity: A mechanistic study. Biochem. Pharmacol. 2019, 159, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Choi, I.S.; Ban, J.O.; Lee, H.J.; Lee, U.S.; Han, S.B.; Jung, J.K.; Kim, Y.H.; Kim, K.H.; Oh, K.W.; et al. 4-O-methylhonokiol attenuated β-amyloid-induced memory impairment through reduction of oxidative damages via inactivation of p38 MAP kinase. J. Nutr. Biochem. 2011, 22, 476–486. [Google Scholar] [CrossRef]
- Choi, I.S.; Lee, Y.J.; Choi, D.Y.; Lee, Y.K.; Lee, Y.H.; Kim, K.H.; Kim, Y.H.; Jeon, Y.H.; Kim, E.H.; Han, S.B.; et al. 4-O-methylhonokiol attenuated memory impairment through modulation of oxidative damage of enzymes involving amyloid-β generation and accumulation in a mouse model of alzheimer’s disease. J. Alzheimer Dis. 2011, 27, 127–141. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, D.Y.; Lee, Y.K.; Lee, Y.M.; Han, S.B.; Kim, Y.H.; Kim, K.H.; Nam, S.Y.; Lee, B.J.; Kang, J.K.; et al. 4-O-methylhonokiol prevents memory impairment in the tg2576 transgenic mice model of alzheimer’s disease via regulation of β-secretase activity. J. Alzheimer Dis. 2012, 29, 677–690. [Google Scholar] [CrossRef]
- Jung, Y.-Y.; Lee, Y.-J.; Choi, D.-Y.; Hong, J.T. Amelioration of Cognitive Dysfunction in APP/PS1 Double Transgenic Mice by Long-Term Treatment of 4-O-Methylhonokiol. Biomol. Ther. 2014, 22, 232–238. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, D.Y.; Choi, I.S.; Kim, K.H.; Kim, Y.H.; Kim, H.M.; Lee, K.; Cho, W.G.; Jung, J.K.; Han, S.B.; et al. Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappaB in vitro and in vivo models. J. Neuroinflamm. 2012, 9, 35. [Google Scholar] [CrossRef]
- Askari, V.R.; Nick, R.S. The protective effects of β-caryophyllene on LPS-induced primary microglia M1/M2 imbalance: A mechanistic evaluation. Life Sci. 2019, 219, 40–73. [Google Scholar] [CrossRef]
- Hu, Y.; Zeng, Z.; Wang, B.; Guo, S. Trans-caryophyllene inhibits amyloid β (Aβ) oligomer-induced neuroinflammation in BV-2 microglial cells. Int. Immunopharmacol. 2017, 51, 91–98. [Google Scholar] [CrossRef]
- Schuehly, W.; Paredes, J.M.V.; Kleyer, J.; Huefner, A.; Goffer, S.A.; Raduner, S.; Altmann, K.-H.; Gertsch, J. Mechanisms of osteoclastogenesis inhibition by a novel class of biphenyl-type cannabinoid CB (2) receptor inverse agonists. Chem. Biol. 2011, 18, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, J.; Ahn, S.; An, S.; Ko, H.; Shin, J.C.; Jin, S.H.; Ki, M.W.; Lee, S.H.; Lee, K.H.; et al. Diallyl biphenyl-type neolignans have a pharmacophore of pparα/γ dual modulators. Biomol. Ther. 2020, 28, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid type 2 (CB2) receptors activation protects against oxidative stress and neuroinflammation associated dopaminergic neurodegeneration in rotenone model of parkinson’s disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.M.V.; Castañeda, R.E.G.; Gertsch, J.; Huerta, V.C.; Roa, R.I.L.; Valls, E.V.; Zarate, C.B.; Espuny, A.C.; Soto, M.E.F. Neuroprotective Effects of β-caryophyllene against dopaminergic neuron injury in a murine model of parkinson’s disease induced by MPTP. Pharmaceuticals 2017, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ma, W.; Du, J. β-Caryophyllene (BCP) ameliorates MPP+ induced cytotoxicity. BioMed Pharmacother. 2018, 103, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Book, S.W. Anxiety and substance use disorders: A review. Psychiatr. Times 2008, 25, 19–23. [Google Scholar] [PubMed]
- Bahi, A.; Mansouri, S.A.; Memari, E.A.; Ameri, M.A.; Nurulain, S.M.; Ojha, S. β-Caryophyllene, a CB2 receptor agonist produces multiple behavioral changes relevant to anxiety and depression in mice. Physiol. Behav. 2014, 135, 119–124. [Google Scholar] [CrossRef]
- Schwarz, R.; Ramer, R.; Hinz, B. Targeting the endocannabinoid system as a potential anticancer approach. Drug Metab. Rev. 2018, 50, 26–53. [Google Scholar] [CrossRef]
- Gómez, E.P.; Andradas, C.; Benito, S.B.; Caffarel, M.M.; Taboada, E.G.; Morales, M.V.; Moreno, E.; Hamann, S.; Villar, E.M.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar]
- McKallip, R.J.; Lombard, C.; Fisher, M.; Martin, B.R.; Ryu, S.; Grant, S.; Nagarkatti, P.S.; Nagarkatti, M. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood 2002, 100, 627–634. [Google Scholar] [CrossRef]
- Sánchez, C.; del Pulgar, T.G.; Rueda, D.; Velasco, G.; Roperh, I.G.; Guzmán, M.; de Ceballos, M.L.; Corbacho, C.; Ramón y Cajal, S.; Huffman, J.W. Inhibition of glioma growth in vivo by selective activation of the CB2 cannabinoid receptor. Cancer Res. 2001, 61, 5784–5789. [Google Scholar] [PubMed]
- Burstein, S.H. Ajulemic acid: Potential treatment for chronic inflammation. Pharmacol. Res. Perspect. 2018, 6, e00394. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Smith, T.W. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015, 5, 362. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Oh, J.; Ban, J.; Shim, J.; Lee, H.; Jung, J.; Ahn, B.; Yoon, D.; Han, S.; Ham, Y.; et al. 4-O-methylhonokiol, a PPARγ agonist, inhibits prostate tumour growth: p21-mediated suppression of NF-κB activity. Br. J. Pharmacol. 2013, 168, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lee, R.H.; Jeon, Y.J.; Shin, J.C.; Park, S.M.; Choi, N.J.; Seo, K.S.; Yoon, G.; Cho, S.S.; Kim, K.H.; et al. Role of transcription factor Sp1 in the 4-O-methylhonokiol-mediated apoptotic effect on oral squamous cancer cells and xenograft. Int. J. Biochem. Cell Biol. 2015, 64, 287–297. [Google Scholar] [CrossRef]
- Xiao, S.; Chen, F.; Gao, C. Antitumor activity of 4-O-Methylhonokiol in human oral cancer cells is mediated via ROS generation, disruption of mitochondrial potential, cell cycle arrest and modulation of Bcl-2/Bax proteins. JBUON 2017, 22, 1577–1581. [Google Scholar]
- Oh, J.H.; Ban, J.O.; Cho, M.C.; Jo, M.; Jung, J.K.; Ahn, B.; Yoon, D.Y.; Han, S.B.; Hong, J.T. 4-O-methylhonokiol inhibits colon tumor growth via p21-mediated suppression of NF-κB activity. J. Nutr. Biochem. 2012, 23, 706–715. [Google Scholar] [CrossRef]
- Hall, J.A.; Rusten, M.; Abughazaleh, R.D.; Wuertz, B.; Souksavong, V.; Escher, P.; Ondrey, F. Effects of PPAR-γ agonists on oral cancer cell lines: Potential horizons for chemopreventives and adjunctive therapies. Head Neck 2020, 42, 2542–2554. [Google Scholar] [CrossRef]
- Ban, J.O.; Kwak, D.H.; Oh, J.H.; Park, E.J.; Cho, M.C.; Song, H.S.; Song, M.J.; Han, S.B.; Moon, D.C.; Kang, K.W.; et al. Suppression of NF-κB and GSK-3β is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chem. Biol. Interact. 2010, 188, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, V.S.; Kaur, G. Insulinotropic and antidiabetic effects of β-caryophyllene with l-arginine in type 2 diabetic rats. J. Food Biochem. 2020, 44, e13156. [Google Scholar] [CrossRef] [PubMed]
- Suijun, W.; Zhen, Y.; Ying, G.; Yanfang, W. A role for trans-caryophyllene in the moderation of insulin secretion. Biochem. Biophys. Res. Commun. 2014, 444, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.A.; Fayoumi, H.M.E.; Mahmoud, M.F. Beta-caryophyllene protects against diet-induced dyslipidemia and vascular inflammation in rats: Involvement of CB2 and PPAR-γ receptors. Chem. Biol. Interact. 2019, 297, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Tharappel, L.J.P.; Kumawat, V. Evaluation of safety and in vitro mechanisms of anti-diabetic activity of β-caryophyllene and L-arginine. J. Biol. Sci. 2018, 18, 124–134. [Google Scholar] [CrossRef]
- Varga, Z.V.; Matyas, C.; Erdelyi, K.; Cinar, R.; Nieri, D.; Chicca, A.; Nemeth, B.T.; Paloczi, J.; Lajtos, T.; Corey, L.; et al. β-Caryophyllene protects against alcoholic steatohepatitis by attenuating inflammation and metabolic dysregulation in mice. Br. J. Pharmacol. 2018, 175, 320–334. [Google Scholar] [CrossRef]
- Scandiffio, R.; Geddo, F.; Cottone, E.; Querio, G.; Antoniotti, S.; Gallo, M.P.; Maffei, M.E.; Bovolin, P. Protective Effects of (E)-β-Caryophyllene (BCP) in Chronic Inflammation. Nutrients 2020, 12, 3273. [Google Scholar] [CrossRef]
- Rossi, F.; Punzo, F.; Umano, G.R.; Argenziano, M.; del Giudice, E.M. Role of cannabinoids in obesity. Int. J. Mol. Sci. 2018, 19, 2690. [Google Scholar] [CrossRef]
- Ma, T.; Zheng, Z.; Guo, H.; Lian, X.; Rane, M.J.; Cai, L.; Kim, K.S.; Kim, K.T.; Zhang, Z.; Bi, L. 4-O-methylhonokiol ameliorates type 2 diabetes-induced nephropathy in mice likely by activation of AMPK-mediated fatty acid oxidation and Nrf2-mediated anti-oxidative stress. Toxicol. Appl. Pharmacol. 2019, 370, 93–105. [Google Scholar] [CrossRef]
- Zheng, Z.; Ma, T.; Guo, H.; Kim, K.S.; Kim, K.T.; Bi, L.; Zhang, Z.; Cai, L. 4-O-methylhonokiol protects against diabetic cardiomyopathy in type 2 diabetic mice by activation of AMPK-mediated cardiac lipid metabolism improvement. J. Cell. Mol. Med. 2019, 23, 5771–5781. [Google Scholar] [CrossRef]
- Patsenker, E.; Chicca, A.; Petrucci, V.; Moghadamrad, S.; de Gottardi, A.; Hampe, J.; Gertsch, J.; Semmo, N.; Stickel, F. 4-O′-methylhonokiol protects from alcohol/carbon tetrachloride-induced liver injury in mice. J. Mol. Med. 2017, 95, 1077–1089. [Google Scholar] [CrossRef]
- Deutsch, D.G.; Chin, S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993, 46, 791–796. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed]
- di Marzo, V.; Melck, D.; Bisogno, T.; de Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998, 21, 521–528. [Google Scholar] [CrossRef]
- Brunetti, L.; Loiodice, F.; Piemontese, L.; Tortorella, P.; Laghezza, A. New Approaches to Cancer Therapy: Combining Fatty Acid Amide Hydrolase (FAAH) Inhibition with Peroxisome Proliferator-Activated Receptors (PPARs) Activation. J. Med. Chem. 2019, 62, 10995–11003. [Google Scholar] [CrossRef] [PubMed]
- Caprioglio, D.; Mattoteia, D.; Pollastro, F.; Negri, R.; Lopatriello, A.; Chianese, G.; Minassi, A.; Collado, J.A.; Munoz, E.; Taglialatela-Scafati, O.; et al. The Oxidation of Phytocannabinoids to Cannabinoquinoids. J. Nat. Prod. 2020, 83, 1711–1715. [Google Scholar] [CrossRef]
- Scafati, O.T.; Pagani, A.; Scala, F.; de Petrocellis, L.; di Marzo, V.; Grassi, G.; Appendino, G. Cannabimovone, a cannabinoid with a rearranged terpenoid skeleton from hemp. Eur. J. Org. Chem. 2010, 2010, 2067–2072. [Google Scholar] [CrossRef]
- Iannotti, F.A.; de Maio, F.; Panza, E.; Appendino, G.; Scafati, O.T.; de Petrocellis, L.; Amodeo, P.; Vitale, R.M. Identification and Characterization of Cannabimovone, a Cannabinoid from Cannabis sativa, as a Novel PPARγ Agonist via a Combined Computational and Functional Study. Molecules 2020, 25, 1119. [Google Scholar] [CrossRef]
- Sanz, G.M. Can You Pass the Acid Test? Critical Review and Novel Therapeutic Perspectives of Δ9-Tetrahydrocannabinolic Acid A. Cannabis Cannabinoid Res. 2016, 1, 124–130. [Google Scholar] [CrossRef]
- Raman, P.; Kaplan, B.L.F.; Thompson, J.T.; Heuvel, J.P.V.; Kaminski, N.E. Metabolite of 2-Arachidonyl Glycerol, Activates Peroxisome Proliferator Activated Receptor gamma. Mol. Pharmacol. 2011, 80, 201–209. [Google Scholar] [CrossRef]
- Fakhrudin, N.; Ladurner, A.; Atanasov, A.G.; Heiss, E.H.; Baumgartner, L.; Markt, P.; Schuster, D.; Ellmerer, E.P.; Wolber, G.; Rollinger, J.M.; et al. Computer-aided discovery, validation, and mechanistic characterization of novel neolignan activators of peroxisome proliferator-activated receptor γ. Mol. Pharmacol. 2010, 77, 559–566. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z.; Huang, X.; Shi, W.; Zhang, R.; Chen, M.; Huang, H.; Wu, L. Insights on the Multifunctional Activities of Magnolol. BioMed Res. Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Lin, M.H.; Chen, M.C.; Chen, T.H.; Chang, H.Y.; Chou, T.C. Magnolol ameliorates lipopolysaccharide-induced acute lung injury in rats through PPAR-γ-dependent inhibition of NF-kB activation. Int. Immunopharmacol. 2015, 28, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Zhang, Z.; He, Y.; Gu, C.; Zhu, K.; Li, S.; Li, Y.; Lu, X.; Liu, J.; Zhang, N.; et al. Magnolol treatment attenuates dextran sulphate sodium-induced murine experimental colitis by regulating inflammation and mucosal damage. Life Sci. 2018, 196, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhao, J.; Wang, H.; Jiang, Y.; Yang, Q.; Fu, Y.; Zeng, H.; Hölscher, C.; Xu, J.; Zhang, Z. Magnolol alleviates Alzheimer’s disease-like pathology in transgenic C. elegans by promoting microglia phagocytosis and the degradation of beta-amyloid through activation of PPAR-γ. BioMed Pharmacother. 2020, 124, 109886. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.Y.; Chou, T.C. The antiplatelet activity of magnolol is mediated by PPAR-β/γ. Biochem. Pharmacol. 2012, 84, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xing, W.; He, J.; Fu, F.; Zhang, W.; Su, F.; Liu, F.; Ji, L.; Gao, F.; Su, H.; et al. Magnolol administration in normotensive young spontaneously hypertensive rats postpones the development of hypertension: Role of increased PPAR gamma, reduced TRB3 and resultant alleviative vascular insulin resistance. PLoS ONE 2015, 10, e0120366. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Cha, B.Y.; Lee, Y.S.; Yonezawa, T.; Teruya, T.; Nagai, K.; Woo, J.T. Magnolol enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Life Sci. 2009, 84, 908–914. [Google Scholar] [CrossRef]
- Rempel, V.; Fuchs, A.; Hinz, S.; Karcz, T.; Lehr, M.; Koetter, U.; Müller, C.E. Magnolia extract, magnolol, and metabolites: Activation of cannabinoid CB2 receptors and blockade of the related GPR55. ACS Med. Chem. Lett. 2013, 4, 41–45. [Google Scholar] [CrossRef]
- Fuchs, A.; Rempel, V.; Müller, C.E. The natural product magnolol as a lead structure for the development of potent cannabinoid receptor agonists. PLoS ONE 2013, 8, e77739. [Google Scholar] [CrossRef]
- D’Aniello, E.; Fellous, T.; Iannotti, F.A.; Gentile, A.; Allarà, M.; Balestrieri, F.; Gray, R.; Amodeo, P.; Vitale, R.M.; Di Marzo, V. Identification and characterization of phytocannabinoids as novel dual PPARα/γ agonists by a computational and in vitro experimental approach. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 586–597. [Google Scholar] [CrossRef]
- Priestley, R.S.; Nickolls, S.A.; Alexander, S.P.H.; Kendall, D.A. A potential role for cannabinoid receptors in the therapeutic action of fenofibrate. FASEB J. 2015, 29, 1446–1455. [Google Scholar] [CrossRef]
- McGuinness, D.; Malikzay, A.; Visconti, R.; Lin, K.; Bayne, M.; Monsma, F.; Lunn, C.A. Characterizing cannabinoid CB2 receptor ligands using DiscoveRx PathHunterTM β-arrestin assay. J. Biomol. Screen. 2009, 14, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Verme, J.L.; Fu, J.; Astarita, G.; Rana, G.L.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.L.B.B.; Dias, S.M.G.G.; Polikarpov, I.; Zurier, R.B.; Burstein, S.H.; Garratt, R.C. Ajulemic acid, a synthetic nonpsychoactive cannabinoid acid, bound to the ligand binding domain of the human peroxisome proliferator-activated receptor. J. Biol. Chem. 2007, 282, 18625–18633. [Google Scholar] [CrossRef]
- Tepper, M.A.; Zurier, R.B.; Burstein, S.H. Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg. Med. Chem. 2014, 22, 3245–3251. [Google Scholar] [CrossRef]
- Martín, A.G.; Rodríguez, M.G.; Navarrete, C.; Caprioglio, D.; Palomares, B.; de Mesa, J.; Rollland, A.; Appendino, G.; Muñoz, E. Cannabinoid derivatives acting as dual PPARγ/CB2 agonists as therapeutic agents for systemic sclerosis. Biochem. Pharmacol. 2019, 163, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, M.D.; Richardson, D.; Robinson, I.; Garle, M.J.; Patel, A.; Sun, Y.; Sagar, D.R.; Bennett, A.J.; Alexander, S.P.H.; Kendall, D.A.; et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology 2008, 55, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. In Phytocannabinoids: Unraveling the Complex Chemistry and Pharmacology of Cannabis Sativa; Kinghorn, A.D., Gibbons, S., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 103, pp. 103–131. ISBN 978-3-319-45539-6. [Google Scholar]
- Trial to Evaluate Efficacy and Safety of Lenabasum in Dermatomyositis. Available online: https://clinicaltrials.gov/ct2/show/NCT03813160?term=lenabasum&draw=2&rank=2 (accessed on 3 November 2020).
- Kroker, A.J.; Bruning, J.B. Review of the structural and dynamic mechanisms of PPAR γ partial agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W.R. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-γ–RXR-α nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef]
- Cronet, P.; Petersen, J.F.W.; Folmer, R.; Blomberg, N.; Sjöblom, K.; Karlsson, U.; Lindstedt, E.L.; Bamberg, K. Structure of the PPARα and -γ ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar] [CrossRef]
- Mileni, M.; Kamtekar, S.; Wood, D.C.; Benson, T.E.; Cravatt, B.F.; Stevens, R.C. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: Discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 2010, 400, 743–754. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Otte, P.S.; Luttrell, L.M.; Martin, B.; Maudsley, S. β-Arrestin based receptor signaling paradigms: Potential therapeutic targets for complex age-related disorders. Front. Pharmacol. 2018, 9, 1369. [Google Scholar] [CrossRef] [PubMed]
- Miljuš, T.; Heydenreich, F.M.; Gazzi, T.; Kimbara, A.; Nettekoven, M.; Zirwes, E.; Osterwald, A.; Rufer, A.C.; Ullmer, C.; Guba, W.; et al. Diverse chemotypes drive biased signaling by cannabinoid. bioRxiv 2020, 375162. [Google Scholar] [CrossRef]
- Zoubi, R.A.; Morales, P.; Reggio, P.H. Structural Insights into CB1 Receptor Biased Signaling. Int. J. Mol. Sci. 2019, 20, 1837. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Goya, P.; Jagerovic, N. Emerging strategies targeting CB2 cannabinoid receptor: Biased agonism and allosterism. Biochem. Pharmacol. 2018, 157, 8–17. [Google Scholar] [CrossRef]
- Kang, J.; Shi, Y.; Xiang, B.; Qu, B.; Su, W.; Zhu, M.; Zhang, M.; Bao, G.; Wang, F.; Zhang, X.; et al. A nuclear function of β-arrestin1 in GPCR signaling: Regulation of histone acetylation and gene transcription. Cell 2005, 123, 833–847. [Google Scholar] [CrossRef]
- Hoeppner, C.Z.; Cheng, N.; Ye, R.D. Identification of a nuclear localization sequence in β-arrestin-1 and its functional implications. J. Biol. Chem. 2012, 287, 8932–8943. [Google Scholar] [CrossRef]
- Wang, C.; Zeng, X.; Zhou, Z.; Zhao, J.; Pei, G. β-arrestin-1 contributes to brown fat function and directly interacts with PPARα and PPARγ. Sci. Rep. 2016, 6, 26999. [Google Scholar] [CrossRef]
- Zhuang, L.N.; Hu, W.X.; Xin, S.M.; Zhao, J.; Pei, G. β-arrestin-1 protein represses adipogenesis and inflammatory responses through its interaction with peroxisome proliferator-activated receptor-γ (PPARγ). J. Biol. Chem. 2011, 286, 28403–28413. [Google Scholar] [CrossRef]
- Gorgulla, C.; Padmanabha Das, K.M.; Leigh, K.E.; Cespugli, M.; Fischer, P.D.; Wang, Z.F.; Tesseyre, G.; Pandita, S.; Shnapir, A.; Calderaio, A.; et al. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. ChemRxiv 2020, 102021. [Google Scholar] [CrossRef]



{kind=link}
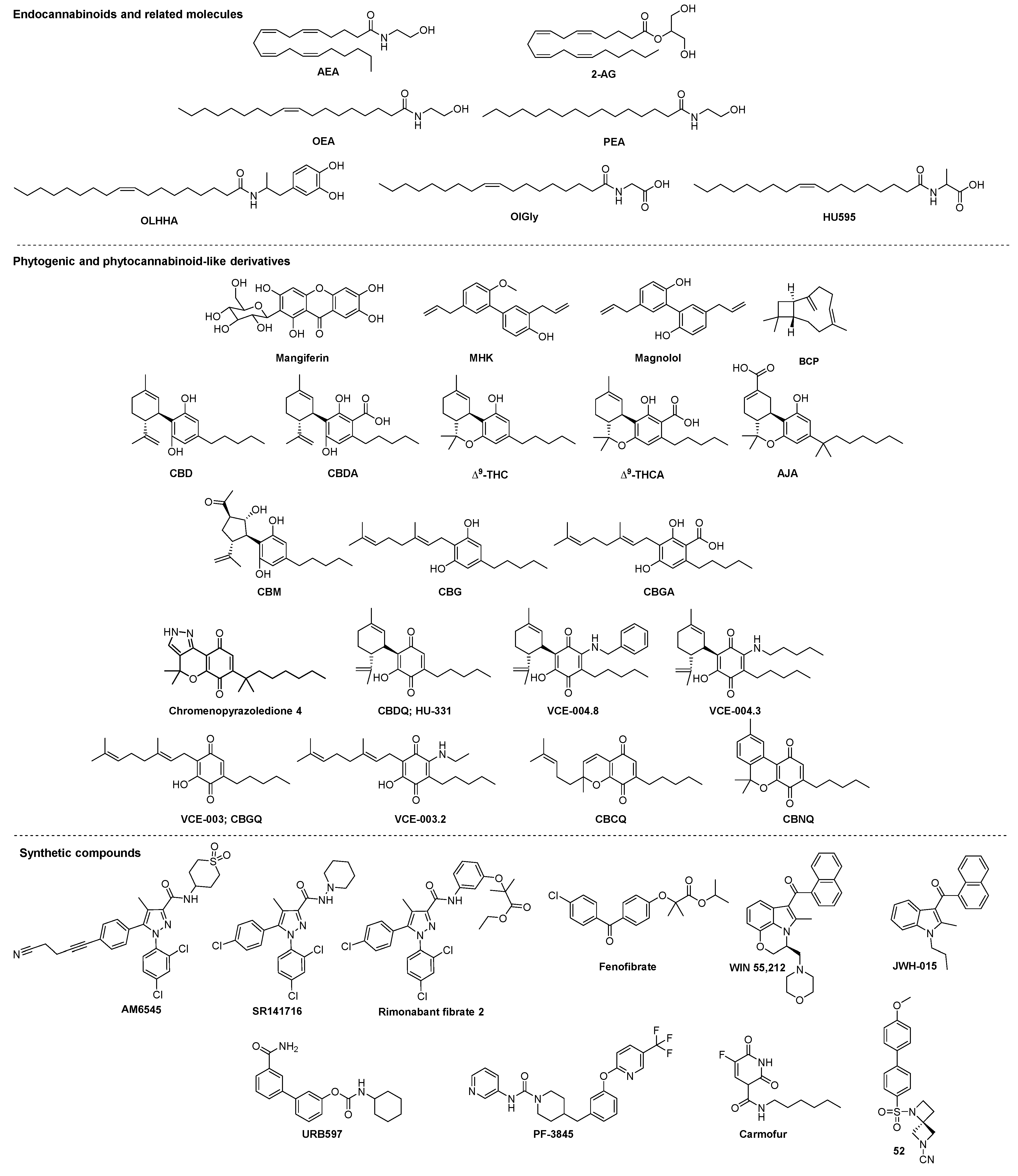{kind=link}
{kind=link}
| Targets Involved | Pathologies | Compounds | Ref. |
|---|---|---|---|
| CB1R–PPARα | Metabolic syndrome | OLHHA | [48,49,50] |
| Rimonabant fibrate 2 | [52] | ||
| AM6545 | [53] | ||
| OEA + SR144716A | [54] | ||
| Alcohol use disorder | OLHHA | [51] | |
| Epilepsy | PEA | [61] | |
| Nociception | PEA | [65] | |
| AEA + GW7647 | [66] | ||
| CB1R–PPARγ | Cancer | Chromenopyrazoledione 4 | [71] |
| CBD | [73] | ||
| Obesity | EMI | [77] | |
| Arthritis | THCA | [81] | |
| CB2R–PPARγ | Arthritis | AJA | [83] |
| BCP | [84] | ||
| Fibrosis | AJA | [85] | |
| VCE-004.3 | [86] | ||
| VCE-004.8 | [87,88] | ||
| Multiple sclerosis | BCP | [89] | |
| VCE-004.8 | [90] | ||
| Alzheimer’s disease | MHK | [91] | |
| BCP | [92] | ||
| Parkinson’s disease | BCP | [93] | |
| Substance abuse | BCP | [94,95] | |
| Cancer | AJA | [96] | |
| MHK | [97] | ||
| BCP | [98] | ||
| WIN55,212-2 | [99] | ||
| JHW-015 | [55] | ||
| Metabolic dysfunction | VCE-004.8 | [100] | |
| MHK | [101] | ||
| CB2R–PPARα | Ulcerative colitis | PEA | [102] |
| CB2R–PPARα/γ | Metabolic dysfunction | BCP | [103] |
| FAAH–PPARα | Inflammation | Carmofur ‡‡ | [104] |
| Azetidine-nitrile 52 | [105] | ||
| Nausea | PF-3845 | [106] | |
| Opioid tolerance | URB597 ‡‡‡ | [107] | |
| Opioid withdrawal | HU595 ‡ | [108] | |
| OlGly ‡ | [109] | ||
| Nicotine withdrawal | OlGly ‡ | [110] | |
| CBR–PPARγ | Memory | CBD * | [111] |
| Alzheimer’s disease | CBD * | [112] | |
| WIN55,212-2 ** | [113] | ||
| Huntington’s disease | THCA* | [82] | |
| VCE-003.2 * | [114,115] | ||
| Parkinson’s disease | VCE-003.2 * | [116,117] | |
| Multiple sclerosis | VCE-003 * | [118] | |
| BBB permeability following ischemia | CBD * | [119] | |
| Vasorelaxation | CBD * | [74] | |
| THC * | [120] | ||
| Addiction | WIN55,212-2 * | [121] | |
| CBR–PPARα | Cancer | THC * | [122] |
| Anticonvulsant | WIN55,212-2 ** | [123] | |
| Inflammation | PEA * | [124] | |
| Nausea | THCA * | [125] |
| Compounds | Targets | References | ||||
|---|---|---|---|---|---|---|
| CB1R | CB2R | PPARα | PPARγ | FAAH | ||
| OEA | + | [56] | ||||
| PEA | [+] | [+] | + | + | [61,65,102] | |
| OLHHA | - | + | [48,50,51] | |||
| OlGly | [+] | NE | + | - | [109,110] | |
| HU595 | [+] | + | - | [106,108] | ||
| Magnolol | + | + | + | + | [159,197,204] | |
| MHK | * | + | + | [159,186] | ||
| EMI | UR | UR | [77] | |||
| BCP | NE | + | + | [+] | [84,139,140] | |
| CBD | * | * | + | [74,111,112,118,119,206] | ||
| CBDA | + | + | [206] | |||
| THC | + | + | * | + | [41,74,120,122] | |
| THCA | + *** | - | [+] | + | [81,82,125,194] | |
| AJA | + | NE | + | [210,211] | ||
| CBM | NE | NE | + | + | [193] | |
| CBG | + | + | [74,191,206] | |||
| CBGA | + | + | [206] | |||
| Chromenopyrazole 4 | + | + | [71] | |||
| VCE-004.8 | NE | + | + | [212] | ||
| VCE-004.3 | - | + | + | [86] | ||
| VCE-003; CBGQ | NE | + | + | [118,191] | ||
| VCE-003.2 | NE | NE | + | [114,115,116,117] | ||
| CBDQ; HU-331 | NE | NE | + | [191] | ||
| CBCQ | + | [191] | ||||
| CBNQ | + | [191] | ||||
| Fenofibrate | + | + | + | [207] | ||
| AM6545 | - ** | [+] | [53] | |||
| Rimonabant fibrate 2 | - | + | [52] | |||
| WIN55,212 | + | + | + | [+] | [41,99,113,123] | |
| JHW-015 | + | [+] | [55] | |||
| URB597 | [+] | [+] | [+] | - | [106,107,213] | |
| PF-3845 | [+] | [+] | - | [107] | ||
| Carmofur | [+] | [+] | - | [104] | ||
| Azetidine-nitrile 52 | [+] | [+] | [+] | - | [105] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lago-Fernandez, A.; Zarzo-Arias, S.; Jagerovic, N.; Morales, P. Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System. Int. J. Mol. Sci. 2021, 22, 1001. https://doi.org/10.3390/ijms22031001
Lago-Fernandez A, Zarzo-Arias S, Jagerovic N, Morales P. Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System. International Journal of Molecular Sciences. 2021; 22(3):1001. https://doi.org/10.3390/ijms22031001
Chicago/Turabian StyleLago-Fernandez, Ana, Sara Zarzo-Arias, Nadine Jagerovic, and Paula Morales. 2021. "Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System" International Journal of Molecular Sciences 22, no. 3: 1001. https://doi.org/10.3390/ijms22031001
APA StyleLago-Fernandez, A., Zarzo-Arias, S., Jagerovic, N., & Morales, P. (2021). Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System. International Journal of Molecular Sciences, 22(3), 1001. https://doi.org/10.3390/ijms22031001