
Cannabinoid Receptors in Myocardial Injury: A Brother Born to Rival


Abstract
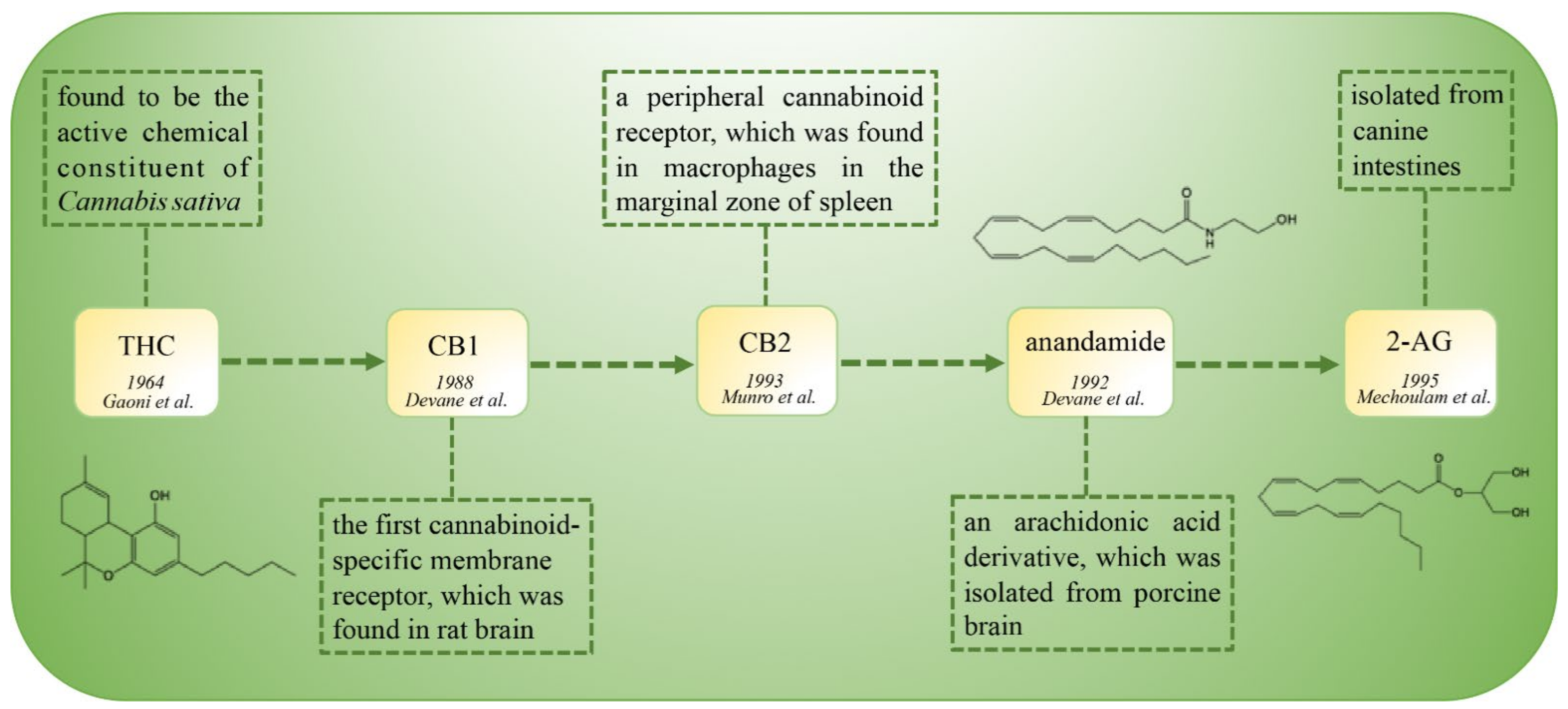
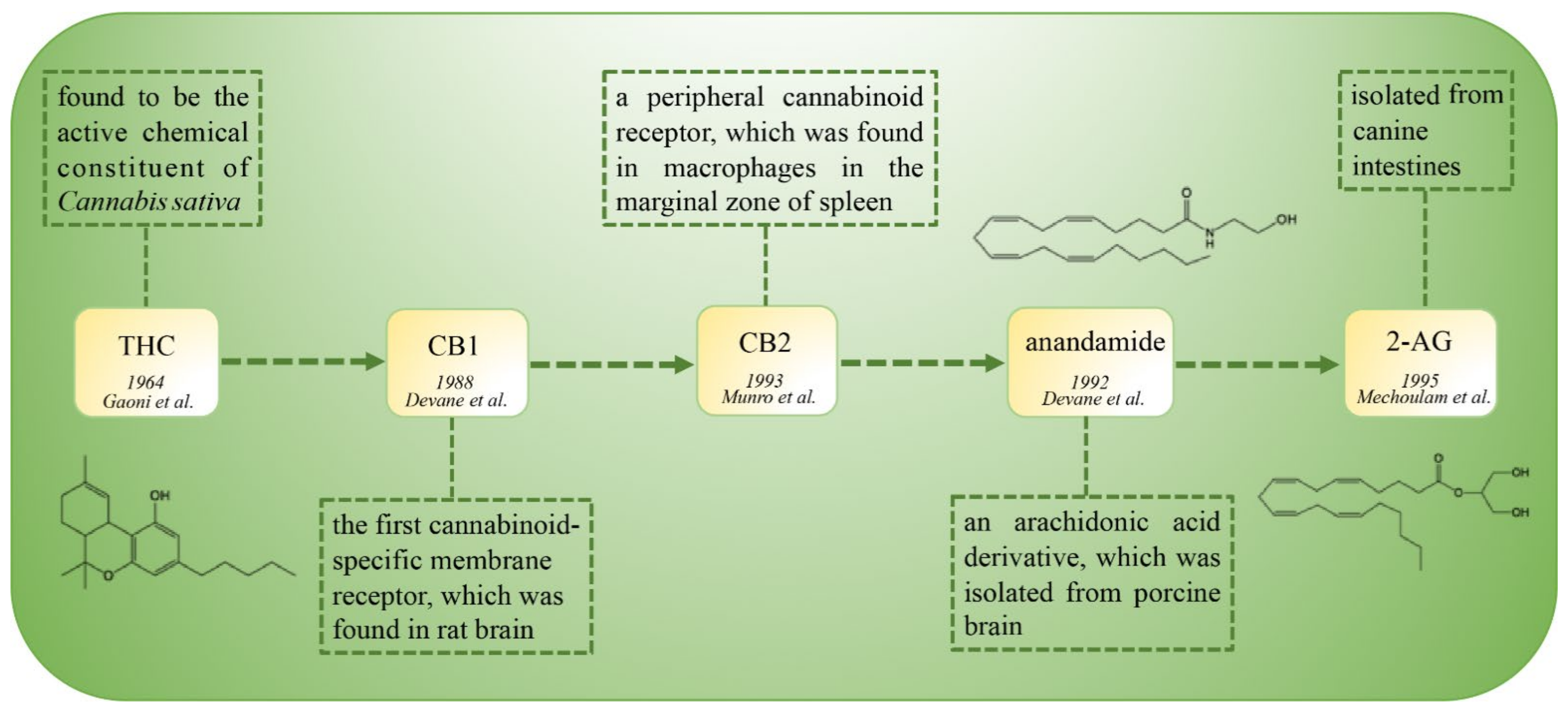
:1. Introduction
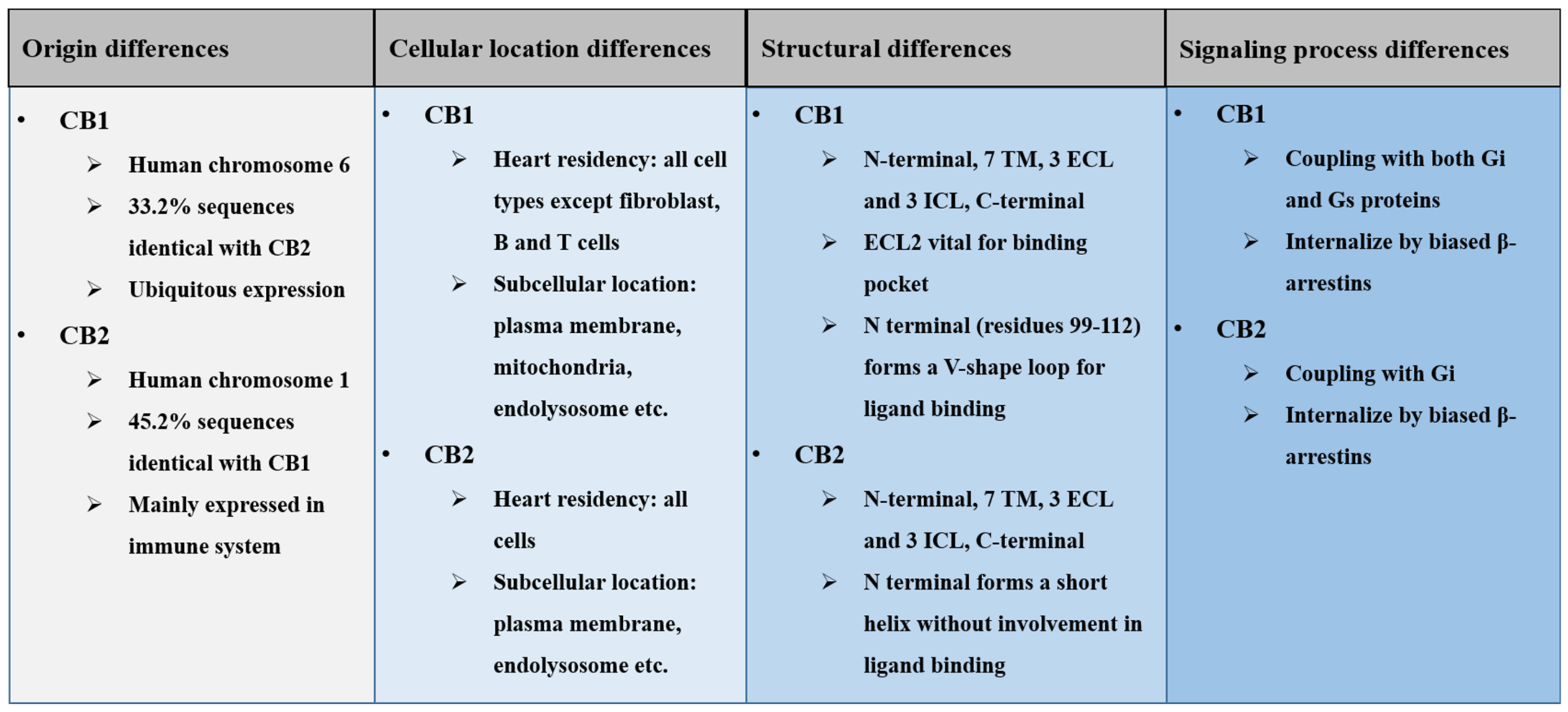
2. General Differences of Cannabinoid Receptors
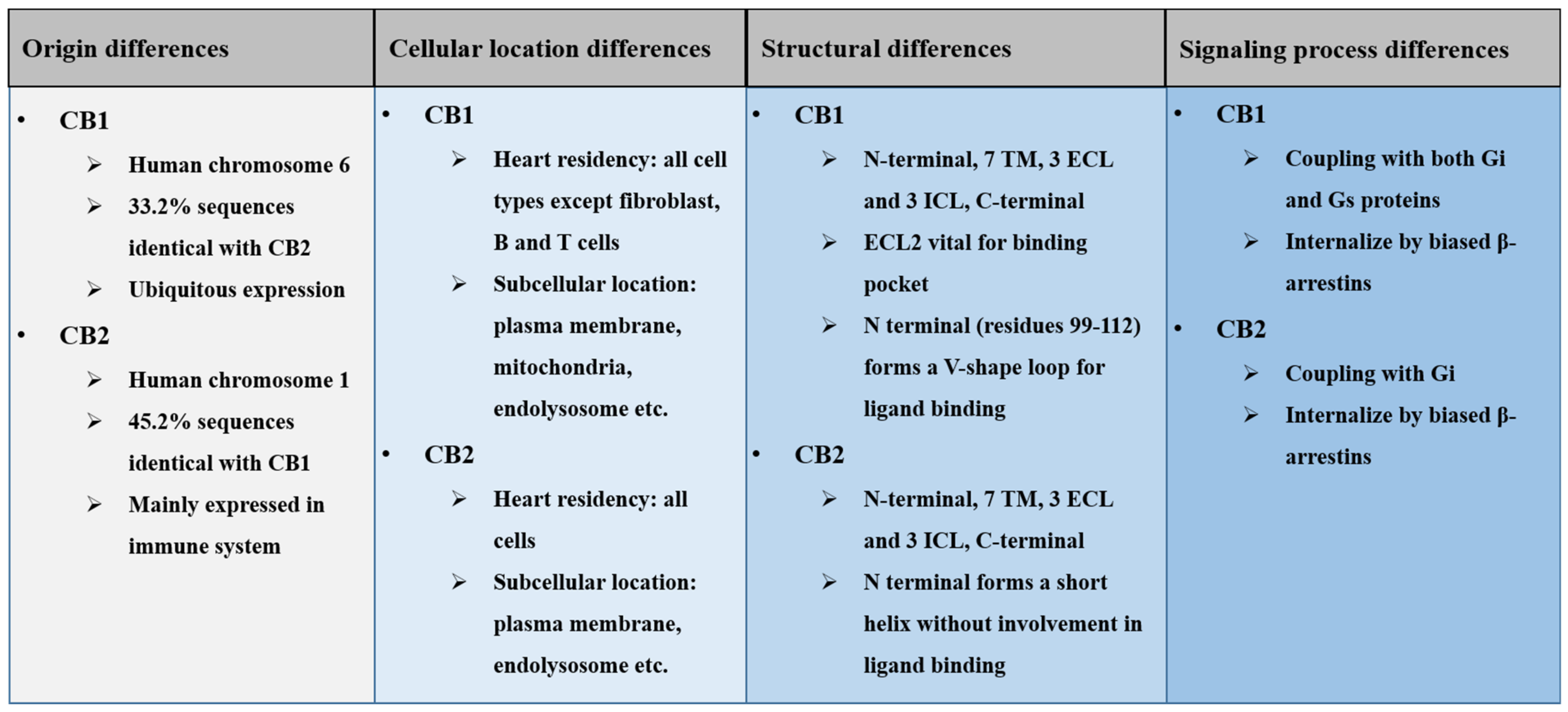
2.1. Origin Differences of Cannabinoid Receptors
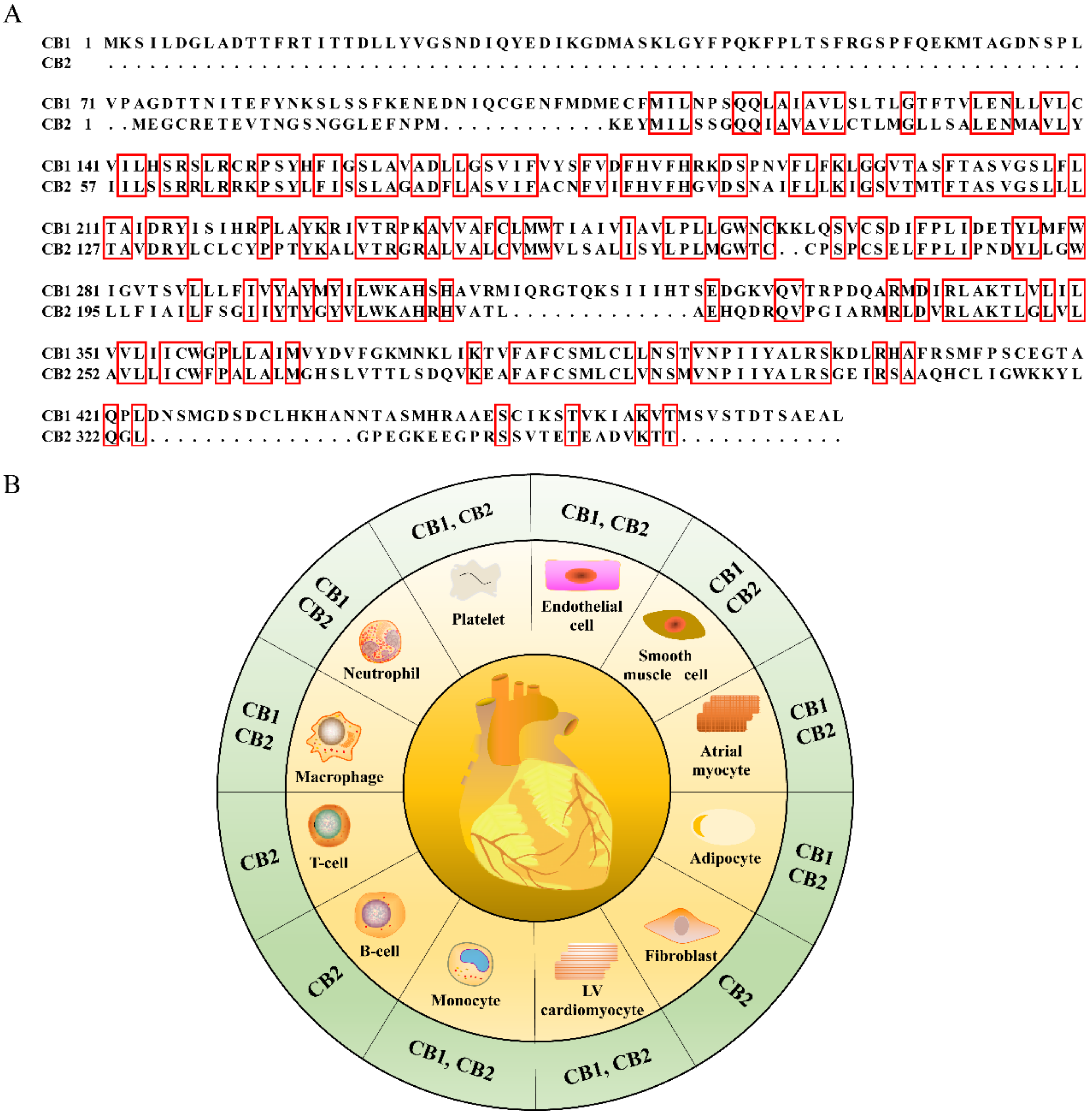
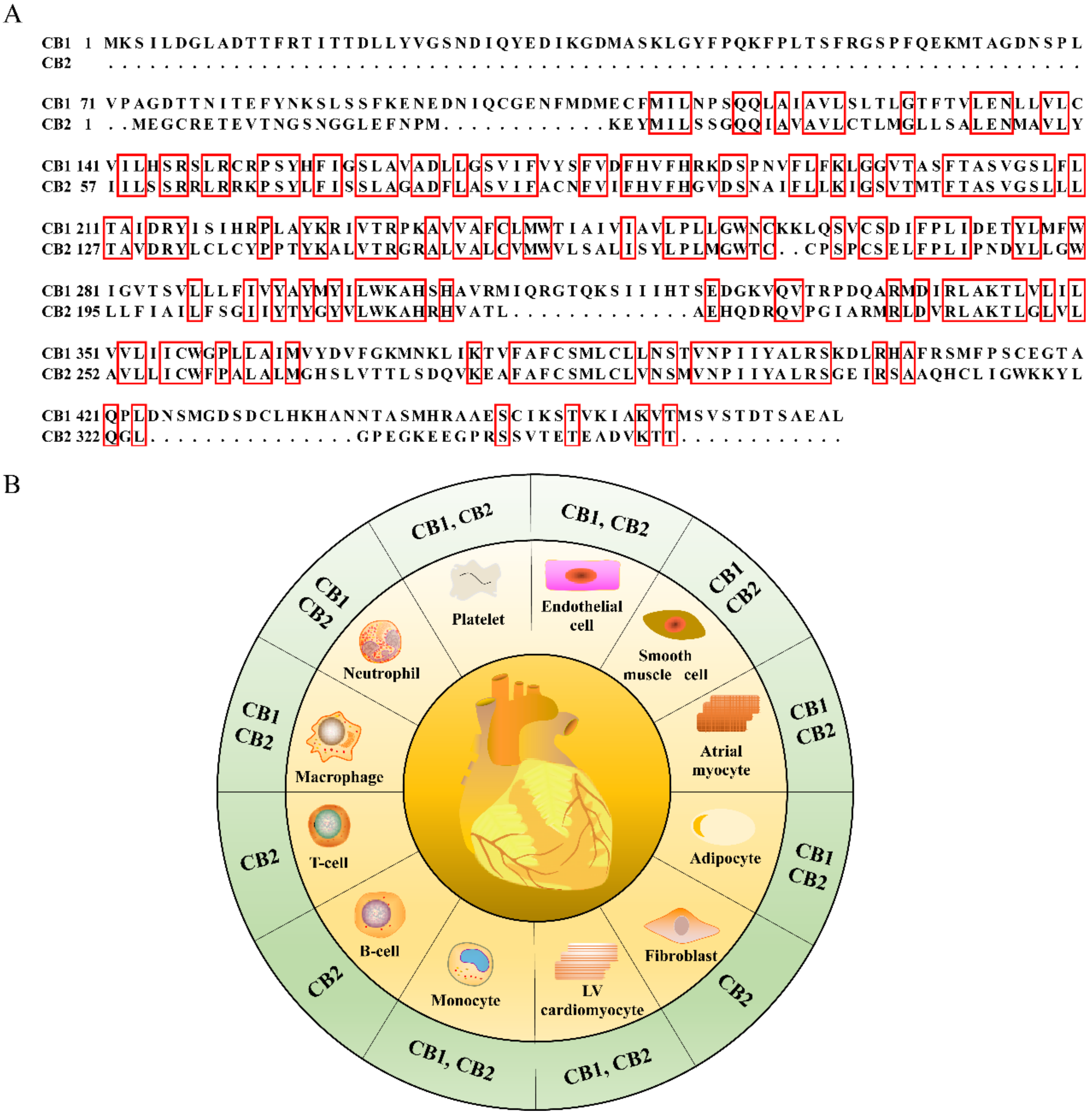
2.2. Cellular Location Differences of Cannabinoid Receptors
2.3. Structural Differences of Cannabinoid Receptors
2.4. Signaling Difference of Cannabinoid Receptors
3. Functional Rivalries between Cannabinoid Receptors in Myocardial Injury
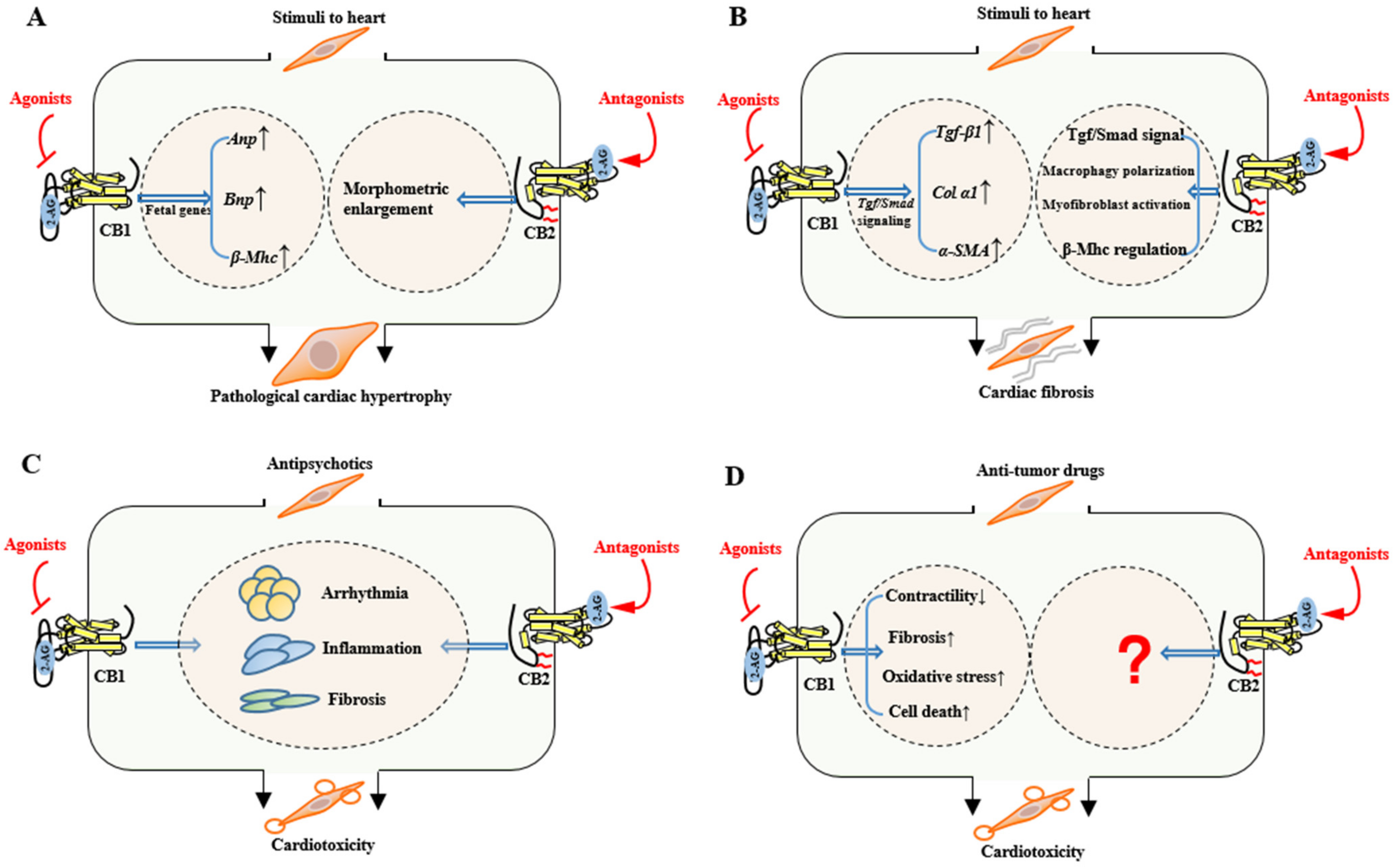
3.1. Acute Myocardial Infarction (MI)
3.2. Cardiac Ischemia/Reperfusion (I/R) Injury
3.3. Pathological Cardiac Hypertrophy
3.4. Cardiac Fibrosis
3.5. Miscellaneous Myocardial Injury
3.5.1. Antipsychotic Cardiotoxicity
3.5.2. Anti-Tumor Drug Cardiotoxicity
3.5.3. Ethanol-Induced Myocardial Injury
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CB1 | cannabinoid receptor type 1 |
| CB2 | cannabinoid receptor type 2 |
| CNS | central nervous system |
| ECS | endocannabinoid system |
| AEA | anandamide |
| 2-AG | 2-arachidonoyl glycerol |
| NADA | N-arachidonoyl-dopamine |
| OAE | virodhamine |
| LPI | lysophosphatidylinositol |
| THC | delta-9 tetrahydrocannabinol |
| GPCRs | G-protein-coupled receptors |
| ECL | extracellular loop |
| ICL | intracellular loop |
| PKA | protein kinase A |
| WT | wild type |
| MI | myocardial infarction |
| I/R | ischemic/reperfusion |
| LPS | lipopolysaccharide |
| α-SMA | α-smooth muscle actin |
| SGAs | second-generation antipsychotics |
| DOX | doxorubicin |
| PRSW | preload-recruitable stroke work |
| ESPVR | end-systolic pressure–volume relation |
| TM | transmembrane |
References
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, A.; Abdulrazzaq, G.M.; Chan, S.L.; Penman, J.; Harvey, J.; Alexander, S.P. Cannabinoid Receptor-Related Orphan G Protein-Coupled Receptors. Adv. Pharmacol. 2017, 80, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Cao, Z.; Wang, W.; Zhou, N. New Insights in Cannabinoid Receptor Structure and Signaling. Curr. Mol. Pharmacol. 2019, 12, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef]
- Bonner, T.; Buckley, N.; Young, A.; Brann, M. Identification of a family of muscarinic acetylcholine receptor genes. Science 1987, 237, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Shahbazi, F.; Grandi, V.; Banerjee, A.; Trant, J.F. Cannabinoids and Cannabinoid Receptors: The Story so Far. iScience 2020, 23, 101301. [Google Scholar] [CrossRef]
- Mackie, K. Cannabinoid Receptors: Where They are and What They do. J. Neuroendocr. 2008, 20, 10–14. [Google Scholar] [CrossRef]
- Lago-Fernandez, A.; Zarzo-Arias, S.; Jagerovic, N.; Morales, P. Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System. Int. J. Mol. Sci. 2021, 22, 1001. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Zottola, A.C.P.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Brailoiu, G.C.; Deliu, E.; Marcu, J.; Hoffman, N.E.; Console-Bram, L.; Zhao, P.; Madesh, M.; Abood, M.E.; Brailoiu, E. Differential Activation of Intracellular versus Plasmalemmal CB2 Cannabinoid Receptors. Biochemistry 2014, 53, 4990–4999. [Google Scholar] [CrossRef] [Green Version]
- Hebert-Chatelain, E.; Reguero, L.; Puente, N.; Lutz, B.; Chaouloff, F.; Rossignol, R.; Piazza, P.-V.; Benard, G.; Grandes, P.; Marsicano, G. Cannabinoid control of brain bioenergetics: Exploring the subcellular localization of the CB1 receptor. Mol. Metab. 2014, 3, 495–504. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [Green Version]
- Fay, J.F.; Dunham, T.D.; Farrens, D.L. Cysteine Residues in the Human Cannabinoid Receptor: Only C257 and C264 Are Required for a Functional Receptor, and Steric Bulk at C386 Impairs Antagonist SR141716A Binding. Biochemistry 2005, 44, 8757–8769. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef] [Green Version]
- Puhl, S.-L. Cannabinoid-sensitive receptors in cardiac physiology and ischaemia. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118462. [Google Scholar] [CrossRef]
- Turu, G.; Hunyady, L. Signal transduction of the CB1 cannabinoid receptor. J. Mol. Endocrinol. 2009, 44, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, M.L.; Thewke, D.P. The Endocannabinoid System and Heart Disease: The Role of Cannabinoid Receptor Type 2. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 34–51. [Google Scholar] [CrossRef]
- Slavic, S.; Lauer, D.; Sommerfeld, M.; Kemnitz, U.R.; Grzesiak, A.; Trappiel, M.; Thoene-Reineke, C.; Baulmann, J.; Paulis, L.; Kappert, K.; et al. Cannabinoid receptor 1 inhibition improves cardiac function and remodelling after myocardial infarction and in experimental metabolic syndrome. J. Mol. Med. 2013, 91, 811–823. [Google Scholar] [CrossRef]
- Pacher, P.; Haskó, G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2008, 153, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Sterin-Borda, L.; Del Zar, C.F.; Borda, E. Differential CB1 and CB2 cannabinoid receptor-inotropic response of rat isolated atria: Endogenous signal transduction pathways. Biochem. Pharmacol. 2005, 69, 1705–1713. [Google Scholar] [CrossRef]
- Lipina, C.; Hundal, H.S. The endocannabinoid system: ‘NO’ longer anonymous in the control of nitrergic signalling? J. Mol. Cell Biol. 2017, 9, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Bátkai, S.; Kechrid, M.; Mukhopadhyay, P.; Lee, W.-S.; Horváth, B.; Holovac, E.; Cinar, R.; Liaudet, L.; Mackie, K.; et al. Cannabinoid 1 Receptor Promotes Cardiac Dysfunction, Oxidative Stress, Inflammation, and Fibrosis in Diabetic Cardiomyopathy. Diabetes 2012, 61, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Schloss, M.J.; Horckmans, M.; Guillamat-Prats, R.; Hering, D.; Lauer, E.; Lenglet, S.; Weber, C.; Thomas, A.; Steffens, S. 2-Arachidonoylglycerol mobilizes myeloid cells and worsens heart function after acute myocardial infarction. Cardiovasc. Res. 2018, 115, 602–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, G.D.; Heinemann, J.C.; Suchan, G.; Kolobara, E.; Wenzel, D.; Geisen, C.; Matthey, M.; Passe-Tietjen, K.; Mahmud, W.; Ghanem, A.; et al. The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic Res. Cardiol. 2014, 109, 425. [Google Scholar] [CrossRef] [PubMed]
- Defer, N.; Wan, J.; Souktani, R.; Escoubet, B.; Perier, M.; Caramelle, P.; Manin, S.; Deveaux, V.; Bourin, M.-C.; Zimmer, A.; et al. The cannabinoid receptor type 2 promotes cardiac myocyte and fibroblast survival and protects against ischemia/reperfusion-induced cardiomyopathy. FASEB J. 2009, 23, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, C.; Rossi, F.; Rossi, S.; D’Amico, M. Cannabinoid CB2 receptor activation reduces mouse myocardial ischemia-reperfusion injury: Involvement of cytokine/chemokines and PMN. J. Leukoc. Biol. 2003, 75, 453–459. [Google Scholar] [CrossRef]
- González, C.; Herradón, E.; Abalo, R.; Vera, G.; Perez-Nievas, B.G.; Leza, J.C.; Martín, M.I.; López-Miranda, V. Cannabinoid/agonist WIN 55,212-2 reduces cardiac ischaemia-reperfusion injury in Zucker diabetic fatty rats: Role of CB2 receptors and iNOS/eNOS. Diabetes/Metab. Res. Rev. 2011, 27, 331–340. [Google Scholar] [CrossRef]
- Lagneux, C.; Lamontagne, D. Involvement of cannabinoids in the cardioprotection induced by lipopolysaccharide. Br. J. Pharmacol. 2001, 132, 793–796. [Google Scholar] [CrossRef] [Green Version]
- Lépicier, P.; Bouchard, J.-F.; Lagneux, C.; Lamontagne, D. Endocannabinoids protect the rat isolated heart against ischaemia. Br. J. Pharmacol. 2003, 139, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Underdown, N.J.; Hiley, C.R.; Ford, W. Anandamide reduces infarct size in rat isolated hearts subjected to ischaemia-reperfusion by a novel cannabinoid mechanism. Br. J. Pharmacol. 2005, 146, 809–816. [Google Scholar] [CrossRef]
- Lim, S.Y.; Davidson, S.M.; Yellon, D.; Smith, C.C.T. The cannabinoid CB1 receptor antagonist, rimonabant, protects against acute myocardial infarction. Basic Res. Cardiol. 2009, 104, 781–792. [Google Scholar] [CrossRef]
- Wagner, J.A.; Hu, K.; Karcher, J.; Bauersachs, J.; Schäfer, A.; Laser, M.; Han, H.; Ertl, G. CB1 cannabinoid receptor antagonism promotes remodeling and cannabinoid treatment prevents endothelial dysfunction and hypotension in rats with myocardial infarction. Br. J. Pharmacol. 2003, 138, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Akinwumi, B.C.; Shao, Z.; Anderson, H.D. Ligand Activation of Cannabinoid Receptors Attenuates Hypertrophy of Neonatal Rat Cardiomyocytes. J. Cardiovasc. Pharmacol. 2014, 64, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Duerr, G.D.; Feißt, A.; Halbach, K.; Verfuerth, L.; Gestrich, C.; Wenzel, D.; Zimmer, A.; Breuer, J.; Dewald, O. CB2-deficiency is associated with a stronger hypertrophy and remodeling of the right ventricle in a murine model of left pulmonary artery occlusion. Life Sci. 2018, 215, 96–105. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Hsu, Y.-J.; Hsu, S.-C.; Chen, Y.; Lee, H.-S.; Lin, S.-H.; Huang, S.-M.; Tsai, C.-S.; Shih, C.-C. CB1 cannabinoid receptor antagonist attenuates left ventricular hypertrophy and Akt-mediated cardiac fibrosis in experimental uremia. J. Mol. Cell. Cardiol. 2015, 85, 249–261. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Eid, B.; Neamatallah, T.; Hanafy, A.; El-Bassossy, H.; Binmahfouz, L.; Aldawsari, H.; Hasan, A.; El-Aziz, G.; Vemuri, K.; Makriyannis, A. Interference with TGFβ1-Mediated Inflammation and Fibrosis Underlies Reno-Protective Effects of the CB1 Receptor Neutral Antagonists AM6545 and AM4113 in a Rat Model of Metabolic Syndrome. Molecules 2021, 26, 866. [Google Scholar] [CrossRef]
- Li, X.; Han, D.; Tian, Z.; Gao, B.; Fan, M.; Li, C.; Li, X.; Wang, Y.; Ma, S.; Cao, F. Activation of Cannabinoid Receptor Type II by AM1241 Ameliorates Myocardial Fibrosis via Nrf2-Mediated Inhibition of TGF-β1/Smad3 Pathway in Myocardial Infarction Mice. Cell. Physiol. Biochem. 2016, 39, 1521–1536. [Google Scholar] [CrossRef]
- Julien, B.; Grenard, P.; Teixeira-Clerc, F.; Van Nhieu, J.T.; Li, L.; Karsak, M.; Zimmer, A.; Mallat, A.; Lotersztajn, S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755. [Google Scholar] [CrossRef]
- Razeghi, P.; Essop, M.F.; Huss, J.M.; Abbasi, S.; Manga, N.; Taegtmeyer, H. Hypoxia-induced switches of myosin heavy chain iso-gene expression in rat heart. Biochem. Biophys. Res. Commun. 2003, 303, 1024–1027. [Google Scholar] [CrossRef]
- Jiang, P.; Wang, L.; Zhang, M.; Zhang, M.; Wang, C.; Zhao, R.; Guan, D. Cannabinoid type 2 receptor manipulates skeletal muscle regeneration partly by regulating macrophage M1/M2 polarization in IR injury in mice. Life Sci. 2020, 256, 117989. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Liu, Z.; Lin, X.; Zhong, F.; Li, S.; Tang, X.; Zhang, Y.; Li, L. Second-generation antipsychotics induce cardiotoxicity by disrupting spliceosome signaling: Implications from proteomic and transcriptomic analyses. Pharmacol. Res. 2021, 170, 105714. [Google Scholar] [CrossRef]
- Wang, J.-F.; Min, J.-Y.; Hampton, T.G.; Amende, I.; Yan, X.; Malek, S.; Abelmann, W.H.; Green, A.I.; Zeind, J.; Morgan, J.P. Clozapine-induced myocarditis: Role of catecholamines in a murine model. Eur. J. Pharmacol. 2008, 592, 123–127. [Google Scholar] [CrossRef]
- Li, L.; Dong, X.; Tu, C.; Li, X.; Peng, Z.; Zhou, Y.; Zhang, D.; Jiang, J.; Burke, A.; Zhao, Z.; et al. Opposite effects of cannabinoid CB 1 and CB 2 receptors on antipsychotic clozapine-induced cardiotoxicity. Br. J. Pharmacol. 2019, 176, 890–905. [Google Scholar] [CrossRef]
- Li, X.; Peng, Z.; Zhou, Y.; Wang, J.; Lin, X.; Dong, X.; Liu, X.; Jiang, J.; Jiang, Y.; Li, L. Quetiapine induces myocardial necroptotic cell death through bidirectional regulation of cannabinoid receptors. Toxicol. Lett. 2019, 313, 77–90. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Patel, V.; Kashiwaya, Y.; Liaudet, L.; Evgenov, O.V.; Mackie, K.; Haskó, G.; Pacher, P. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 2009, 85, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Bátkai, S.; Rajesh, M.; Czifra, N.; Harvey-White, J.; Haskó, G.; Zsengeller, Z.; Gerard, N.P.; Liaudet, L.; Kunos, G.; et al. Pharmacological Inhibition of CB1Cannabinoid Receptor Protects Against Doxorubicin-Induced Cardiotoxicity. J. Am. Coll. Cardiol. 2007, 50, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Solà, J. The Effects of Ethanol on the Heart: Alcoholic Cardiomyopathy. Nutrients 2020, 12, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, D.; Dong, X.; Zhu, R.; Ye, Y.; Li, L.; Jiang, Y. Pharmacological activation of CB2 receptor protects against ethanol-induced myocardial injury related to RIP1/RIP3/MLKL-mediated necroptosis. Mol. Cell. Biochem. 2020, 474, 1–14. [Google Scholar] [CrossRef]
- Gary-Bobo, M.; ElAchouri, G.; Scatton, B.; Le Fur, G.; Oury-Donat, F.; Bensaid, M. The Cannabinoid CB1 Receptor Antagonist Rimonabant (SR141716) Inhibits Cell Proliferation and Increases Markers of Adipocyte Maturation in Cultured Mouse 3T3 F442A Preadipocytes. Mol. Pharmacol. 2005, 69, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, T.; Kondo, S.; Kishimoto, S.; Miyashita, T.; Nakane, S.; Kodaka, T.; Suhara, Y.; Takayama, H.; Waku, K. Evidence That 2-Arachidonoylglycerol but Not N-Palmitoylethanolamine or Anandamide Is the Physiological Ligand for the Cannabinoid CB2 Receptor. J. Biol. Chem. 2000, 275, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Duerr, G.D.; Heinemann, J.C.; Dunkel, S.; Zimmer, A.; Lutz, B.; Lerner, R.; Roell, W.; Mellert, F.; Probst, C.; Esmailzadeh, B.; et al. Myocardial hypertrophy is associated with inflammation and activation of endocannabinoid system in patients with aortic valve stenosis. Life Sci. 2013, 92, 976–983. [Google Scholar] [CrossRef]
- Van Nieuwenhoven, F.; Turner, N. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2008, 81, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Bazwinsky-Wutschke, I.; Zipprich, A.; Dehghani, F. Endocannabinoid System in Hepatic Glucose Metabolism, Fatty Liver Disease, and Cirrhosis. Int. J. Mol. Sci. 2019, 20, 2516. [Google Scholar] [CrossRef] [Green Version]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Ai, W.; Zhang, Y.; Tang, Q.-Z.; Yan, L.; Bian, Z.-Y.; Liu, C.; Huang, H.; Bai, X.; Yin, L.; Li, H. Silibinin attenuates cardiac hypertrophy and fibrosis through blocking EGFR-dependent signaling. J. Cell. Biochem. 2010, 110, 1111–1122. [Google Scholar] [CrossRef]
- Liu, L.; Jin, X.; Hu, C.-F.; Zhang, Y.-P.; Zhou, Z.; Li, R.; Shen, C.-X. Amphiregulin enhances cardiac fibrosis and aggravates cardiac dysfunction in mice with experimental myocardial infarction partly through activating EGFR-dependent pathway. Basic Res. Cardiol. 2018, 113, 12. [Google Scholar] [CrossRef]
- Zheng, R.-H.; Zhang, W.-W.; Ji, Y.-N.; Bai, X.-J.; Yan, C.-P.; Wang, J.; Bai, F.; Zhao, Z.-Q. Exogenous supplement of glucagon like peptide-1 protects the heart against aortic banding induced myocardial fibrosis and dysfunction through inhibiting mTOR/p70S6K signaling and promoting autophagy. Eur. J. Pharmacol. 2020, 883, 173318. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, D.; Chen, Y.; Zhan, K.; Meng, Q.; Zhang, X.; Zhu, L.; Yao, X. Si-Miao-Yong-An Decoction attenuates isoprenaline-induced myocardial fibrosis in AMPK-driven Akt/mTOR and TGF-β/SMAD3 pathways. Biomed. Pharmacother. 2020, 130, 110522. [Google Scholar] [CrossRef]
- Traynor, K. FDA advisers wary of expanding quetiapine use: Clinicians air concerns about metabolic effects, tardive dyskinesia. Am. J. Health Pharm. 2009, 66, 880–882. [Google Scholar] [CrossRef]
- Wu, C.-S.; Tsai, Y.; Tsai, H. Antipsychotic Drugs and the Risk of Ventricular Arrhythmia and/or Sudden Cardiac Death: A Nation-wide Case-Crossover Study. J. Am. Hear. Assoc. 2015, 4, e001568. [Google Scholar] [CrossRef]
- Sun, D.; Li, L.; Zhang, X.; Blanchard, T.G.; Fowler, D.R.; Li, L. Causes of Sudden Unexpected Death in Schizophrenia Patients: A forensic autopsy population study. Am. J. Forensic Med. Pathol. 2019, 40, 312–317. [Google Scholar] [CrossRef]
- Li, L.; Ye, X.; Zhao, Z.; Gao, P.; Jiang, Y. Overlooked fatal infectious diseases after long-term antipsychotic use in patients with psychiatric illness. Schizophr. Res. 2018, 195, 258–259. [Google Scholar] [CrossRef]
- Ray, W.A.; Chung, C.P.; Murray, K.T.; Hall, K.; Stein, C.M. Atypical Antipsychotic Drugs and the Risk of Sudden Cardiac Death. N. Engl. J. Med. 2009, 360, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Pucci, M.; Zaplatic, E.; Di Bonaventura, M.M.; Di Bonaventura, E.M.; De Cristofaro, P.; Maccarrone, M.; Cifani, C.; D’Addario, C. On the Role of Central Type-1 Cannabinoid Receptor Gene Regulation in Food Intake and Eating Behaviors. Int. J. Mol. Sci. 2021, 22, 398. [Google Scholar] [CrossRef]
- Topol, E.J.; Bousser, M.-G.; Fox, K.A.; Creager, M.A.; Despres, J.-P.; Easton, J.D.; Hamm, C.W.; Montalescot, G.; Steg, P.G.; Pearson, T.A.; et al. Rimonabant for prevention of cardiovascular events (CRESCENDO): A randomised, multicentre, placebo-controlled trial. Lancet 2010, 376, 517–523. [Google Scholar] [CrossRef]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–558. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Colan, S.D.; Gelber, R.D.; Perez-Atayde, A.R.; Sallan, S.E.; Sanders, S. Late Cardiac Effects of Doxorubicin Therapy for Acute Lymphoblastic Leukemia in Childhood. N. Engl. J. Med. 1991, 324, 808–815. [Google Scholar] [CrossRef]
- Pipicz, M.; Demján, V.; Sárközy, M.; Csont, T. Effects of Cardiovascular Risk Factors on Cardiac STAT3. Int. J. Mol. Sci. 2018, 19, 3572. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, A.; Kleinerman, E.S. Anthracycline-Induced Cardiotoxicity: Causes, Mechanisms, and Prevention. Adv. Exp. Med. Biol. 2020, 1257, 181–192. [Google Scholar] [CrossRef]
- Adamcova, M.; Skarkova, V.; Seifertova, J.; Rudolf, E. Cardiac Troponins are Among Targets of Doxorubicin-Induced Cardiotoxicity in hiPCS-CMs. Int. J. Mol. Sci. 2019, 20, 2638. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | CB1 Function | CB2 Function | References |
|---|---|---|---|
| Myocardial infarction | CB1 aggravated cardiac ischemic injuries | CB2 mitigated cardiac ischemic injuries | [20,23,26,27,28] |
| Cardiac I/R injury | Majority of the literature documents CB1 as a mediator of I/R injury, although there is some controversy across studies | CB2 potently protected from I/R injury | [29,30,31,32,33,34,35,36,37] |
| Pathological cardiac hypertrophy | Majority of the literature documents CB1 as a pro-hypertrophic receptor, and CB1 tended to be not as potent as CB2 in controlling hypertrophy | CB2 potently conferred anti-hypertrophic property | [38,39,40] |
| Cardiac fibrosis | CB1 promoted fibrogenesis mainly through TGF-β1/Smad3 pathway | CB2 ameliorated cardiac fibrosis via TGFβ1-dependent and independent manners | [23,27,29,30,40,41,42,43,44,45,46] |
| Antipsychotics cardiotoxicity | Pharmacological inhibition of CB1 was cardioprotective | Pharmacological activation of CB2 was cardioprotective | [47,48,49,50] |
| Anti-tumor drug cardiotoxicity | Genetic ablation or pharmacological antagonism of CB1 was cardioprotective | Unknown | [51,52] |
| Ethanol-induced myocardial injury | Less known | CB2 attenuated ethanol toxicity | [53,54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.; Liu, Z.; Li, X.; Wang, J.; Li, L. Cannabinoid Receptors in Myocardial Injury: A Brother Born to Rival. Int. J. Mol. Sci. 2021, 22, 6886. https://doi.org/10.3390/ijms22136886
Tang X, Liu Z, Li X, Wang J, Li L. Cannabinoid Receptors in Myocardial Injury: A Brother Born to Rival. International Journal of Molecular Sciences. 2021; 22(13):6886. https://doi.org/10.3390/ijms22136886
Chicago/Turabian StyleTang, Xinru, Zheng Liu, Xiaoqing Li, Jing Wang, and Liliang Li. 2021. "Cannabinoid Receptors in Myocardial Injury: A Brother Born to Rival" International Journal of Molecular Sciences 22, no. 13: 6886. https://doi.org/10.3390/ijms22136886
APA StyleTang, X., Liu, Z., Li, X., Wang, J., & Li, L. (2021). Cannabinoid Receptors in Myocardial Injury: A Brother Born to Rival. International Journal of Molecular Sciences, 22(13), 6886. https://doi.org/10.3390/ijms22136886