
MicroRNAs Regulate Cell Cycle and Cell Death Pathways in Glioblastoma

Abstract
:1. Introduction
2. MiRNAs and Their Role in Cancer Development
2.1. MiRNA: Structure and Biogenesis
2.2. MiRNA: Link to Cancer Development
2.3. MiRNA: Link to GBM
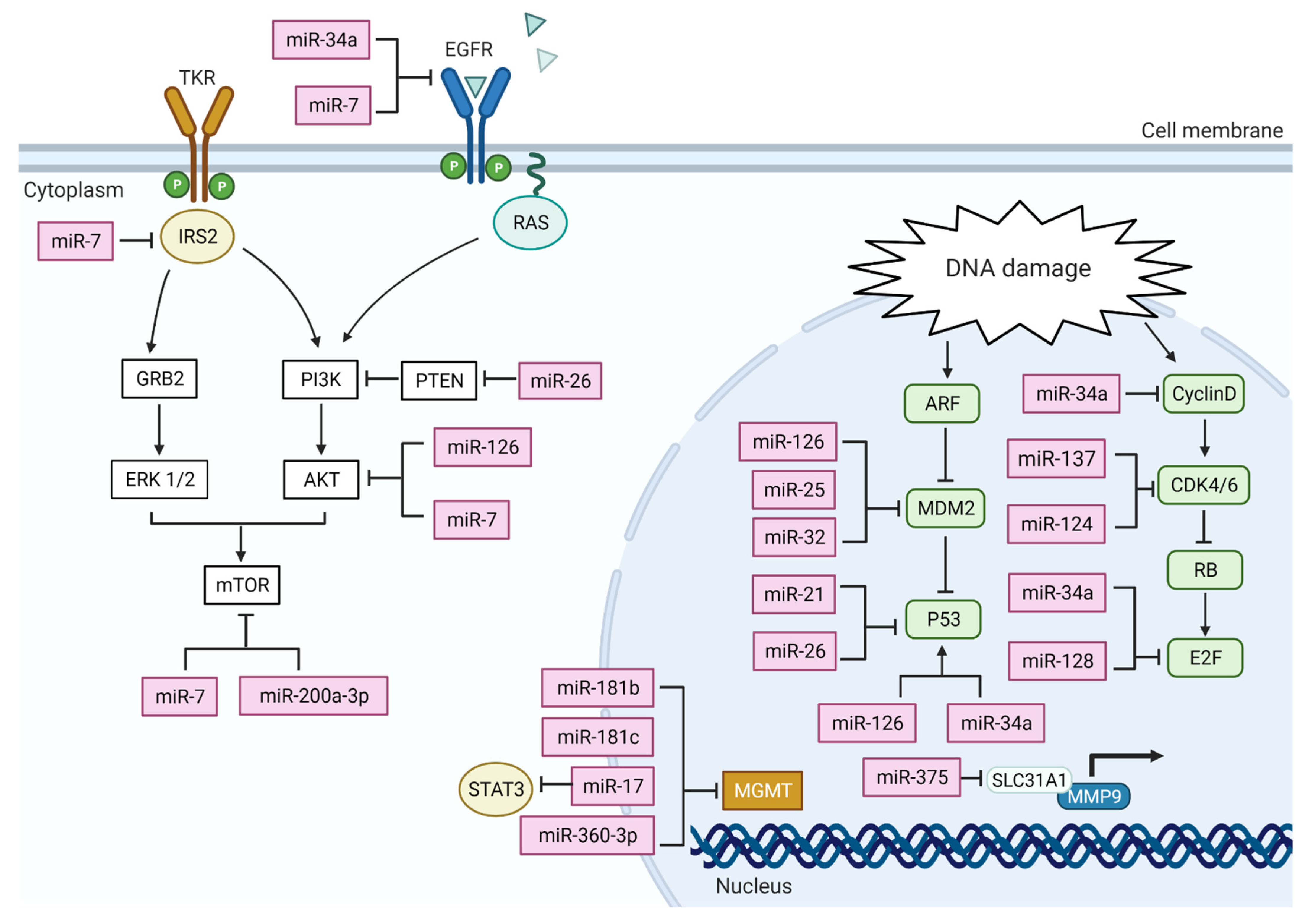
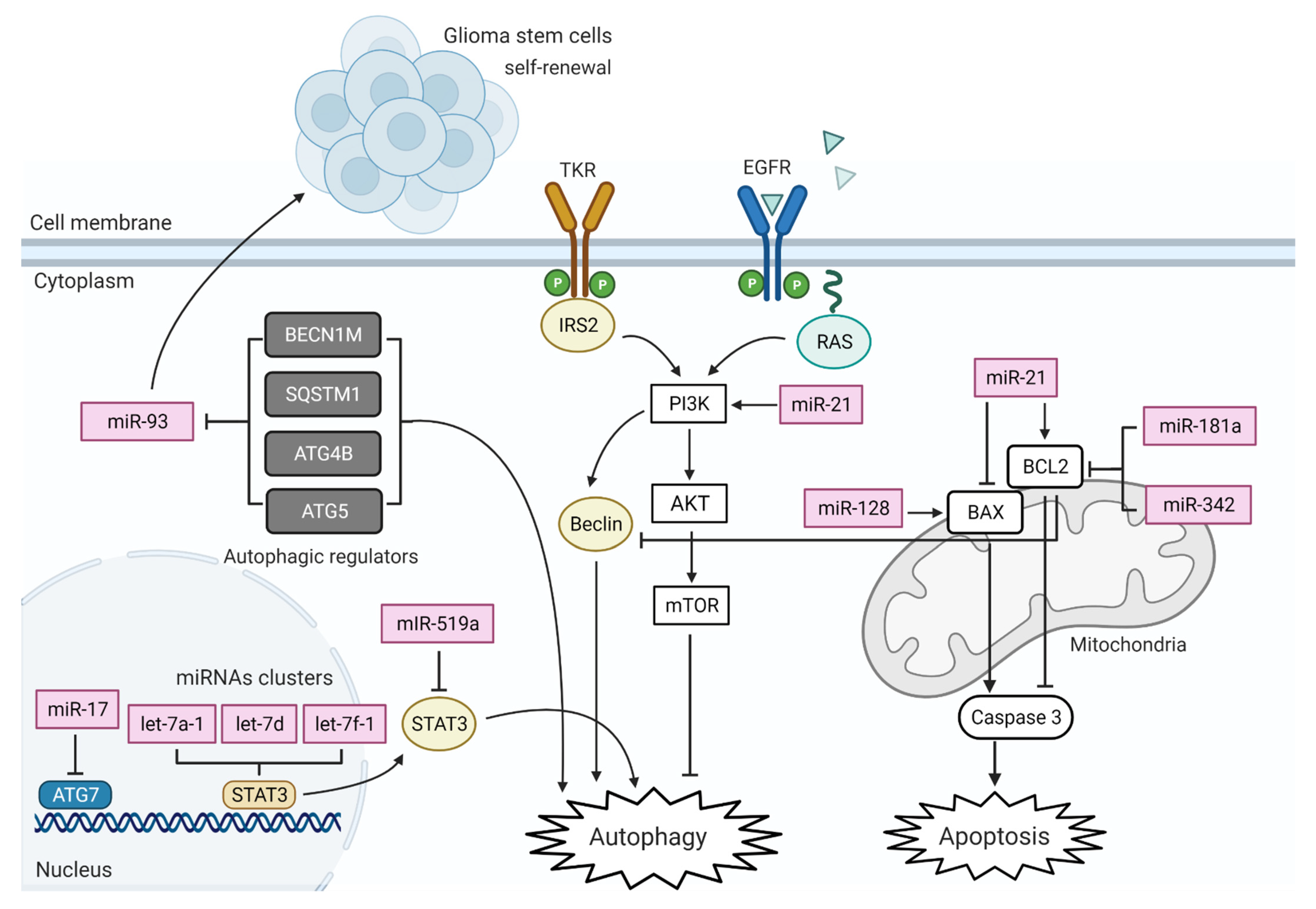
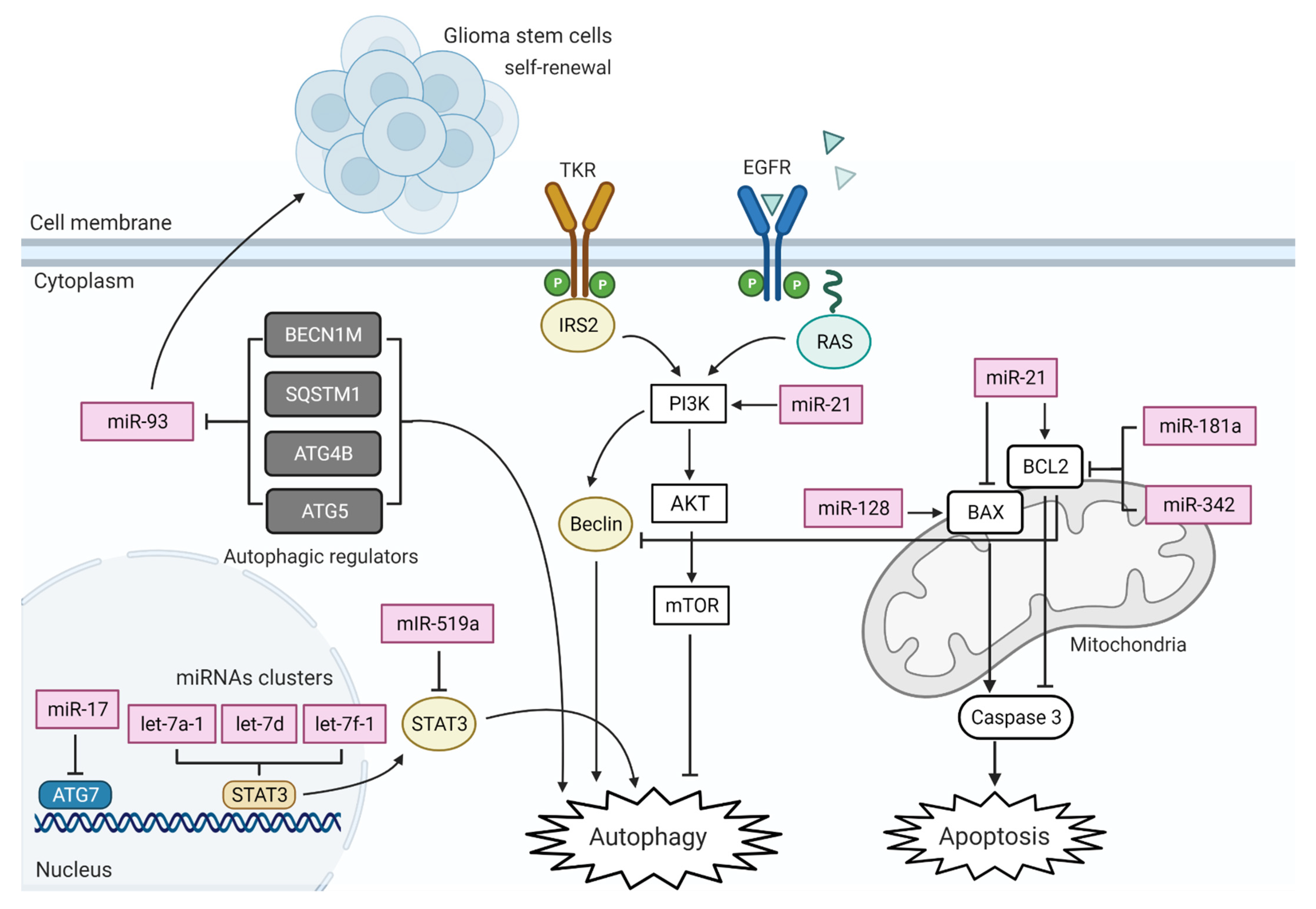
3. MiRNA Actions on GBM Altered Pathways
4. MiRNA in GBM Cell Cycle Control
4.1. Deregulation of P53 Pathway
4.2. Deregulation of Retinoblastoma (RB) Pathway
5. MiRNA Action on PI3K-Akt Pathway
6. MiRNA in GBM Cell Death Pathways
6.1. MiRNA in GBM Mitochondria-Mediated Apoptosis
6.2. Deregulation of Autophagy
6.3. Deregulation of DNA Repair System
7. MiRNA in GBM Inflammation
8. Free Circulating MiRNAs as GBM Diagnostic Tool
9. Possible Use of miRNAs to Predict GBM Treatment Efficiency
10. Viability of Targeting miRNAs in GBM Clinical Settings
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schü, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma: A Population-Based Study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. BioMed Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino, M.D.C.; Otero, A.; Lopes, M.C.; De Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Tachibana, O.; Sato, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223. [Google Scholar] [CrossRef]
- Babashah, S.; Soleimani, M. The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. Eur. J. Cancer 2011, 47, 1127–1137. [Google Scholar] [CrossRef]
- Banelli, B.; Forlani, A.; Allemanni, G.; Morabito, A.; Pistillo, M.P.; Romani, M. MicroRNA in Glioblastoma: An Overview. Int. J. Genom. 2017, 2017, 7639084. [Google Scholar] [CrossRef] [Green Version]
- Lawler, S.; Chiocca, E.A. Emerging functions of microRNAs in glioblastoma. J. Neurooncol. 2009, 92, 297–306. [Google Scholar] [CrossRef]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The roles of microRNAs in breast cancer. Cancers 2015, 7, 598–616. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- González-Gómez, P.; Sánchez, P.; Mira, H. MicroRNAs as regulators of neural stem cell-related pathways in glioblastoma multiforme. Mol. Neurobiol. 2011, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, F.L. MicroRNA and cancer. Mol. Biol. 2014, 48, 232–242. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Tsang, J.S.; Ebert, M.S.; van Oudenaarden, A. Genome-wide Dissection of MicroRNA Functions and Cotargeting Networks Using Gene Set Signatures. Mol. Cell 2010, 38, 140–153. [Google Scholar] [CrossRef] [Green Version]
- O’Carroll, D.; Schaefer, A. General principals of miRNA biogenesis and regulation in the brain. Neuropsychopharmacology 2013, 38, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs Modulate Hematopoietic Lineage Differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esau, C.; Kang, X.; Peralta, E.; Hanson, E.; Marcusson, E.G.; Ravichandran, L.V.; Sun, Y.; Koo, S.; Perera, R.J.; Jain, R.; et al. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem. 2004, 279, 52361–52365. [Google Scholar] [CrossRef] [Green Version]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; MacDonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Liu, C.; Ferracin, M.; Hyslop, T.; Spizzo, R.; Sevignani, C.; Fabbri, M.; Cimmino, A.; Lee, E.J.; Wojcik, S.E.; et al. Ultraconserved Regions Encoding ncRNAs Are Altered in Human Leukemias and Carcinomas. Cancer Cell 2007, 12, 215–229. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; Le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23, 1327–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.-T.; Lin, X.-L.; Liu, Y.; Han, L.-X.; Li, J.; Lin, T.-Y.; Shi, J.-W.; Wang, S.-C.; Lian, M.; Chen, H.-W.; et al. miR-26a promotes hepatocellular carcinoma invasion and metastasis by inhibiting PTEN and inhibits cell growth by repressing EZH2. Lab. Investig. 2019, 99, 1484–1500. [Google Scholar] [CrossRef] [PubMed]
- Miska, E.A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005, 15, 563–568. [Google Scholar] [CrossRef]
- Heinrich, E.M.; Dimmeler, S. MicroRNAs and stem cells: Control of pluripotency, reprogramming, and lineage commitment. Circ. Res. 2012, 110, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Ctor Herranz, H.; Cohen, S.M. MicroRNAs and gene regulatory networks: Managing the impact of noise in biological systems. Genes Dev. 2010, 24, 1339–1344. [Google Scholar] [CrossRef] [Green Version]
- Krichevsky, A.M.; King, K.S.; Donahue, C.P.; Khrapko, K.; Kosik, K.S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003, 9, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Miska, E.A.; Alvarez-Saavedra, E.; Townsend, M.; Yoshii, A.; Sestan, N.; Rakic, P.; Constantine-Paton, M.; Horvitz, H.R. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004, 5, R68. [Google Scholar] [CrossRef] [Green Version]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008, 6, 14. [Google Scholar] [CrossRef]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef] [Green Version]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008, 68, 8164–8172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitjean, A.; Achatz, M.I.W.; Borresen-dale, A.L.; Hainaut, P.; Olivier, M.; Paolo, S. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.J.; Huang, M.S.; Hong, C.Y.; Tse, V.; Silverberg, G.D.; Hsiao, M. Comparisons of tumor suppressor p53, p21, and p16 gene therapy effects on glioblastoma tumorigenicity in Situ. Biochem. Biophys. Res. Commun. 2001, 287, 173–180. [Google Scholar] [CrossRef]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent Co-alterations of TP53, p16/CDKN2A, p14(ARF), PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.R.; Cai, W.P.; Dai, X.J.; Guo, A.S.; Chen, H.P.; Lin, G.S.; Lin, R.S. Research on miR-126 in glioma targeted regulation of PTEN/PI3K/Akt and MDM2-p53 pathways. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3461–3470. [Google Scholar] [CrossRef]
- Suh, S.S.; Yoo, J.Y.; Nuovo, G.J.; Jeon, Y.J.; Kim, S.; Lee, T.J.; Kim, T.; Bakacs, A.; Alder, H.; Kaur, B.; et al. MicroRNAs/TP53 feedback circuitry in glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2012, 109, 5316–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yunqing, L.; Guessous, F.; Ying, Z.; DiPierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jinmai, J.; Yanzhi, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Fu, H.; Liu, Q.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008, 582, 1564–1568. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.; Ogawa, S.; Kawamata, N.; Leiter, A.; Ham, M.; Li, D.; Doan, N.B.; Said, J.W.; Black, K.L.; Phillip Koeffler, H. MiR-34a functions as a tumor suppressor modulating EGFR in glioblastoma multiforme. Oncogene 2013, 32, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Dotto, G.P. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat. Rev. Cancer 2009, 9, 587–595. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. Rb function in cell-cycle regulation and apoptosis. Nat. Cell Biol. 2000, 2, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; RayChaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by MicroRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chao, T.; Li, R.; Liu, W.; Chen, Y.; Yan, X.; Gong, Y.; Yin, B.; Liu, W.; Qiang, B.; et al. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J. Mol. Med. 2009, 87, 43–51. [Google Scholar] [CrossRef]
- Koul, D. PTEN signaling pathways in glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Gwak, H.S.; Kim, T.H.; Jo, G.H.; Kim, Y.J.; Kwak, H.J.; Kim, J.H.; Yin, J.; Yoo, H.; Lee, S.H.; Park, J.B. Silencing of MicroRNA-21 Confers Radio-Sensitivity through Inhibition of the PI3K/AKT Pathway and Enhancing Autophagy in Malignant Glioma Cell Lines. PLoS ONE 2012, 7, e47449. [Google Scholar] [CrossRef]
- Luan, Y.; Zuo, L.; Zhang, S.; Wang, G.; Peng, T. MicroRNA-126 acts as a tumor suppressor in glioma cells by targeting insulin receptor substrate 1 (IRS-1). Int. J. Clin. Exp. Pathol. 2015, 8, 10345–10354. [Google Scholar]
- Cheng, Z.; Luo, C.; Guo, Z. LncRNA-XIST/microRNA-126 sponge mediates cell proliferation and glucose metabolism through the IRS1/PI3K/Akt pathway in glioma. J. Cell. Biochem. 2020, 121, 2170–2183. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-Type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro Oncol. 2017, 19, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Mei, J.; Ji, W.; Huo, Z.; Bian, Z.; Jiao, J.; Li, X.; Sun, J.; Shao, J. MicroRNAs involved in the EGFR pathway in glioblastoma. Biomed. Pharmacother. 2021, 134, 111115. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, Q.; Wang, Y.; Huang, K.; Yang, C.; Li, Y.; Yi, K.; Kang, C. EGFR/c-myc axis regulates TGFβ/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 2017, 406, 12–21. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Z.; Huang, J.; Huang, S.; Li, Y.; Yu, S.; Yu, S.; Liu, X. miR-7 inhibits glioblastoma growth by simultaneously interfering with the PI3K/ATK and Raf/MEK/ERK pathways. Int. J. Oncol. 2014, 44, 1571–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. microRNA-7 inhibits the epidermal growth factor receptor and the akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.M.; Huang, W.; Park, R.; Park, P.J.; Johnson, M.D. A developmental taxonomy of glioblastoma defined and maintained by microRNAs. Cancer Res. 2011, 71, 3387–3399. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Chen, M.B.; Cheng, L.; Zhang, Z.Q.; Yu, Z.Q.; Jiang, Q.; Chen, G.; Cao, C. MicroRNA-200a downregulation in human glioma leads to Gαi1 over-expression, Akt activation, and cell proliferation. Oncogene 2018, 37, 2890–2902. [Google Scholar] [CrossRef]
- Cao, C.; Huang, X.; Han, Y.; Wan, Y.; Birnbaumer, L.; Feng, G.S.; Marshall, J.; Jiang, M.; Chu, W.M. Gαi1 and Gai3 are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci. Signal. 2009, 2, ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartigue, L.; Kushnareva, Y.; Seong, Y.; Lin, H.; Faustin, B.; Newmeyer, D.D. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol. Biol. Cell 2009, 20, 4871–4884. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Zhi, F.; Peng, Y.; Yang, C.C. MiR-128 promotes the apoptosis of glioma cells via binding to NEK2. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8781–8788. [Google Scholar] [CrossRef]
- Chen, G.; Zhu, W.; Shi, D.; Lv, L.; Zhang, C.; Liu, P.; Hu, W. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol. Rep. 2010, 23, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Ciafrè, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, T.C.A.; Bjerkvig, R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer Ther. 2012, 12, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy—A double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Chahal, M.; Abdulkarim, B.; Xu, Y.; Guiot, M.-C.; Easaw, J.C.; Stifani, N.; Sabri, S. Preclinical Development O(6)-Methylguanine-DNA Methyltransferase Is a Novel Negative Effector of Invasion in Glioblastoma Multiforme. Mol. Cancer Ther. 2012, 11, 2440–2450. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaby, O.; Lakomy, R.; Fadrus, P.; Hrstka, R.; Kren, L.; Lzicarova, E.; Smrcka, M.; Svoboda, M.; Dolezalova, H.; Novakova, J.; et al. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma 2010, 57, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahir, B.K.; Ozer, H.; Engelhard, H.H.; Lakka, S.S. Critical Reviews in Oncology/Hematology MicroRNAs in glioblastoma pathogenesis and therapy: A comprehensive review. Crit. Rev. Oncol./Hematol. 2017, 120, 22–33. [Google Scholar] [CrossRef]
- Shi, L.; Chen, J.; Yang, J.; Pan, T.; Zhang, S.; Wang, Z. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010, 1352, 255–264. [Google Scholar] [CrossRef]
- Deng, S.Z.; Lai, M.F.; Li, Y.P.; Xu, C.H.; Zhang, H.R.; Kuang, J.G. Human marrow stromal cells secrete microRNA-375-containing exosomes to regulate glioma progression. Cancer Gene Ther. 2020, 27, 203–215. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Sowers, J.L.; Johnson, K.M.; Conrad, C.; Patterson, J.T.; Sowers, L.C. The role of inflammation in brain cancer. Adv. Exp. Med. Biol. 2014, 816, 75–105. [Google Scholar] [CrossRef] [PubMed]
- Al-kharboosh, R.; ReFaey, K.; Lara-Velazquez, M.; Grewal, S.S.; Imitola, J.; Quiñones-Hinojosa, A. Inflammatory Mediators in Glioma Microenvironment Play a Dual Role in Gliomagenesis and Mesenchymal Stem Cell Homing: Implication for Cellular Therapy. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Hirschberger, S.; Hinske, L.C.; Kreth, S. MiRNAs: Dynamic regulators of immune cell functions in inflammation and cancer. Cancer Lett. 2018, 431, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bandara, K.V.; Michael, M.Z.; Gleadle, J.M. MicroRNA Biogenesis in Hypoxia. MicroRNA 2017, 6, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013, 64, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhou, H.; Xu, J.; Lu, Y.; Ji, X.; Yao, Y.; Chao, H.; Zhang, J.; Zhang, X.; Yao, S.; et al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. 2021, 496, 134–143. [Google Scholar] [CrossRef]
- Hübner, M.; Moellhoff, N.; Effinger, D.; Hinske, C.L.; Hirschberger, S.; Wu, T.; Müller, M.B.; Strauß, G.; Kreth, F.-W.; Kreth, S. MicroRNA-93 acts as an “anti-inflammatory tumor suppressor” in glioblastoma. Neuro-Oncol. Adv. 2020, 2, vdaa047. [Google Scholar] [CrossRef]
- Saadatpour, L.; Fadaee, E.; Fadaei, S.; Nassiri Mansour, R.; Mohammadi, M.; Mousavi, S.M.; Goodarzi, M.; Verdi, J.; Mirzaei, H. Glioblastoma: Exosome and microRNA as novel diagnosis biomarkers. Cancer Gene Ther. 2016, 23, 415–418. [Google Scholar] [CrossRef]
- Roth, P.; Wischhusen, J.; Happold, C.; Chandran, P.A.; Hofer, S.; Eisele, G.; Weller, M.; Keller, A. A specific miRNA signature in the peripheral blood of glioblastoma patients. J. Neurochem. 2011, 118, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M.Y.T.; Young, P.E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M.H.; Shivalingam, B.; et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis. Oncol. 2018, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E. GBM treatment overview. Clin. J. Oncol. Nurs. 2016, 20, S2. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Gong, A.; Yang, H.; George, S.K.; Jiao, Z.; Huang, H.; Jiang, X.; Zhang, Y. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44+/Musashi-1+ gastric cancer stem cells. Cancer Lett. 2015, 369, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Ujifuku, K.; Mitsutake, N.; Takakura, S.; Matsuse, M.; Saenko, V.; Suzuki, K.; Hayashi, K.; Matsuo, T.; Kamada, K.; Nagata, I.; et al. MiR-195, miR-455-3p and miR-10a* are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010, 296, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Shea, A.; Harish, V.; Afzal, Z.; Chijioke, J.; Kedir, H.; Dusmatova, S.; Roy, A.; Ramalinga, M.; Harris, B.; Blancato, J.; et al. MicroRNAs in glioblastoma multiforme pathogenesis and therapeutics. Cancer Med. 2016, 5, 1917–1946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The Risks of miRNA Therapeutics: In a Drug Target Perspective. Drug Des. Dev. Ther. 2021, 15, 721. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| MiRNA | Pathway Affected | Target Gene/s | Expression | Reference |
|---|---|---|---|---|
| mir-137 | CDKs-RB-E2F | CDK6 | downregulated | [41,58] |
| mir-124 | CDKs-RB-E2F | CDK6 | downregulated | [41,58] |
| mir-34a | CDKs-RB-E2F | CyclinD1, cyclinE2, CDK4/6 | downregulated | [54] |
| mir-128 | CDKs-RB-E2F | E2F3a | downregulated | [59] |
| mir-375 | SLC31A1-MMP9 | SLC31A1 | downregulated | [90] |
| mir-21 | ARF-MDM2-P53 | P53 | upregulated | [42] |
| mir-26 | ARF-MDM2-P53 | P53 | up-regulated | [34] |
| mir-126 | ARF-MDM2-P53 | MDM2-P53 | downregulated | [51] |
| mir-126 | PTEN-PI3K-Akt | P13K-Akt | downregulated | [51] |
| mir-25 | ARF-MDM2-P53 | MDM2-TSC1 | downregulated | [52] |
| mir-32 | ARF-MDM2-P53 | MDM2-TSC1 | downregulated | [52] |
| mir-34a | ARF-MDM2-P53 | P53 | downregulated | [53,54,55] |
| mir-34a | Notch pathway | c Met, Notch | downregulated | [53,54,55] |
| mir-21 | Mitochondrial apoptosis | Caspase 3, Bax/BCl2 | upregulated | [89] |
| mir-181b | DNA repair | MGMT | downregulated | [86] |
| mir-181c | DNA repair | MGMT | downregulated | [86] |
| mir-17 | DNA repair | STAT3- MGMT | upregulated | [87] |
| mir-370-3p | DNA repair | MGMT | upregulated | [88] |
| mir-7 | EGFR | EGFR | downregulated | [69] |
| mir-7 | PTEN-PI3K-Akt | P13K- AKT | downregulated | [69] |
| mir-26 | PTEN-PI3K-Akt | PTEN | up-regulated | [34] |
| mir-34a | EGFR | EGFR | downregulated | [55] |
| mir-200a-3p | PTEN-PI3K-Akt | Gαi1-AKT | downregulated | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sati, I.S.E.E.; Parhar, I. MicroRNAs Regulate Cell Cycle and Cell Death Pathways in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 13550. https://doi.org/10.3390/ijms222413550
Sati ISEE, Parhar I. MicroRNAs Regulate Cell Cycle and Cell Death Pathways in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(24):13550. https://doi.org/10.3390/ijms222413550
Chicago/Turabian StyleSati, Isra Saif Eldin Eisa, and Ishwar Parhar. 2021. "MicroRNAs Regulate Cell Cycle and Cell Death Pathways in Glioblastoma" International Journal of Molecular Sciences 22, no. 24: 13550. https://doi.org/10.3390/ijms222413550
APA StyleSati, I. S. E. E., & Parhar, I. (2021). MicroRNAs Regulate Cell Cycle and Cell Death Pathways in Glioblastoma. International Journal of Molecular Sciences, 22(24), 13550. https://doi.org/10.3390/ijms222413550