
Physiological, Biochemical and Transcriptomic Analysis of the Aerial Parts (Leaf-Blade and Petiole) of Asarum sieboldii Responding to Drought Stress


Abstract
:1. Introduction
2. Results
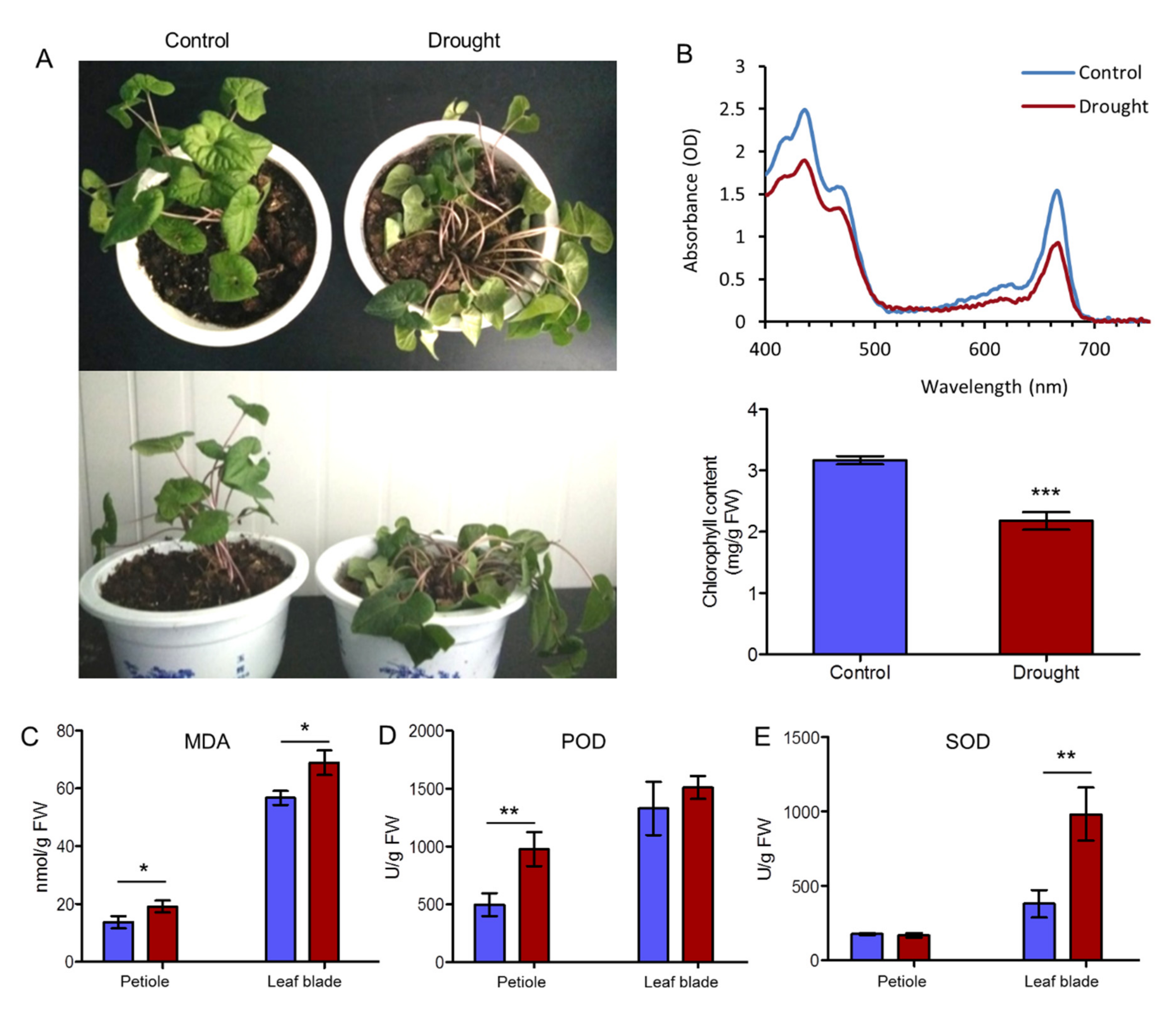
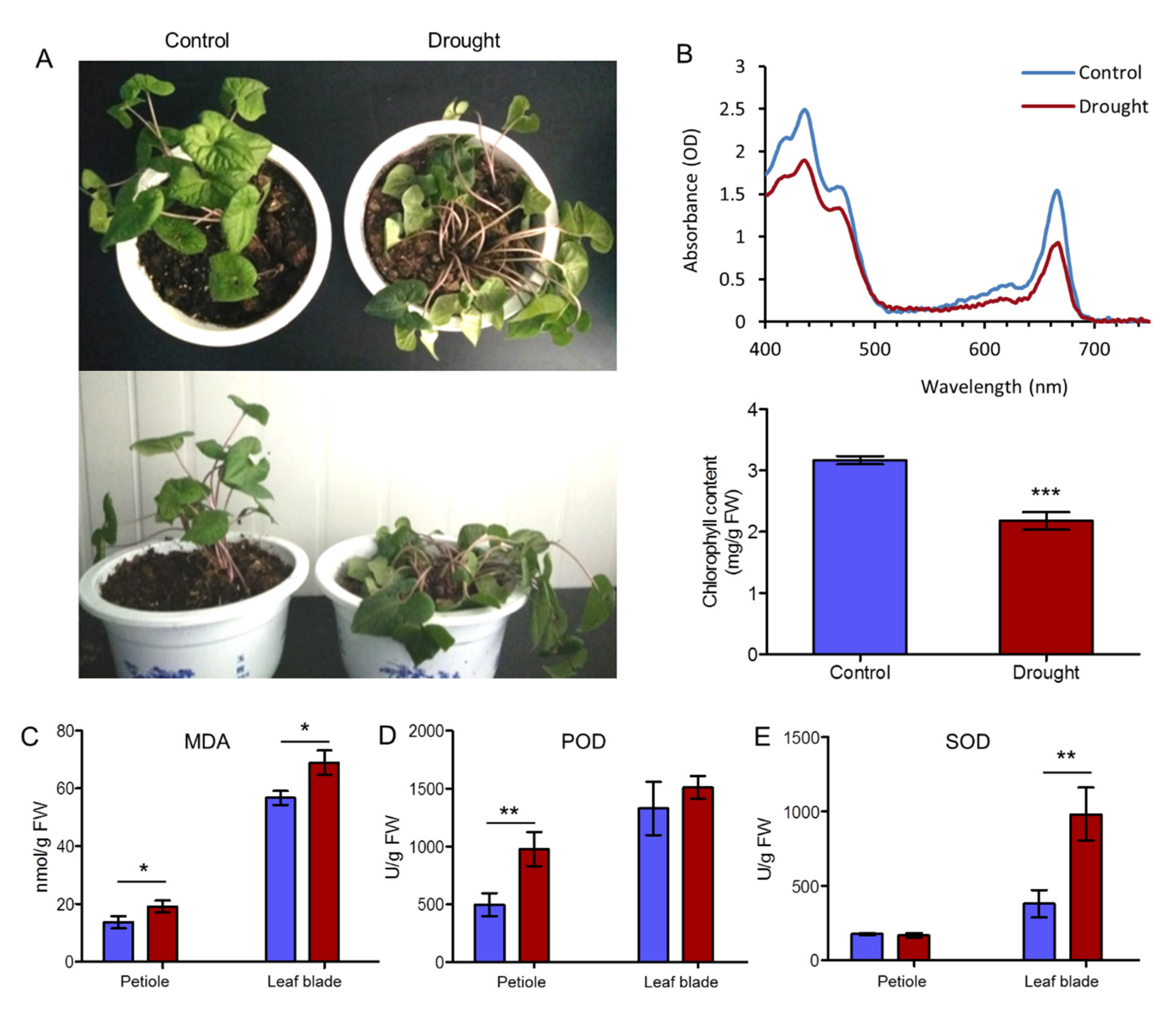
2.1. Physiological Evaluation of A. sieboldii in Response to Drought
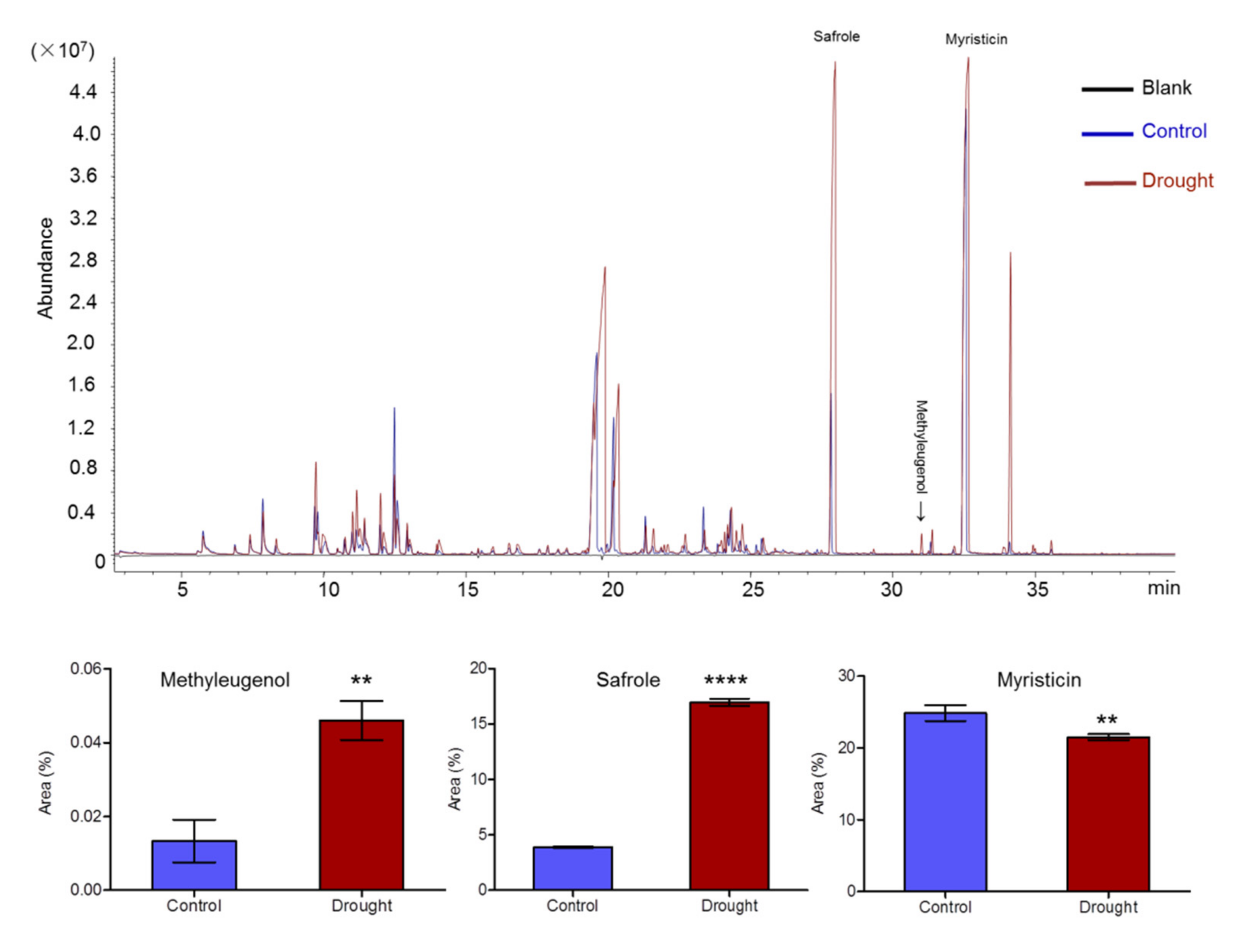
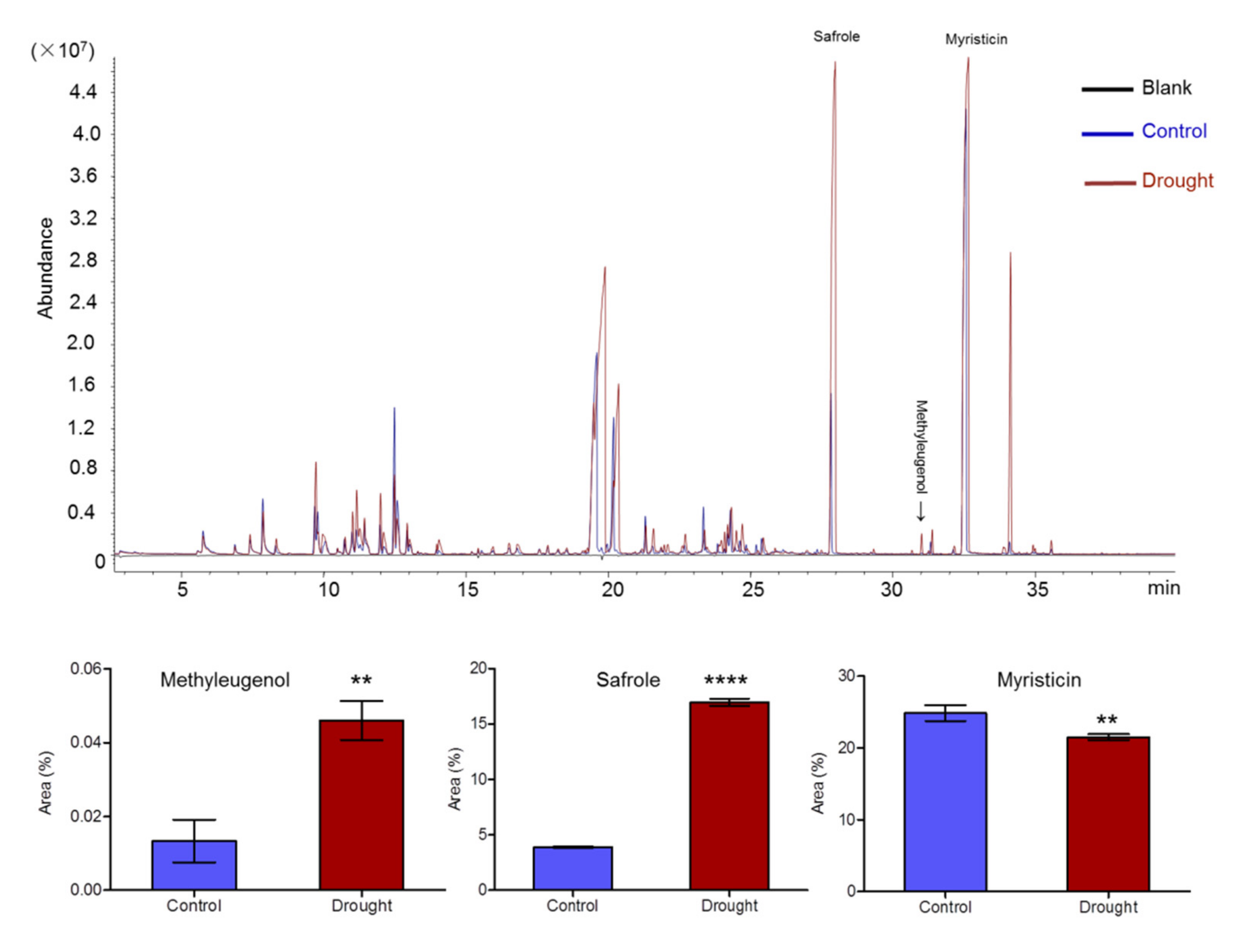
2.2. Essential Oil Accumulation in A. sieboldii under Drought Conditions
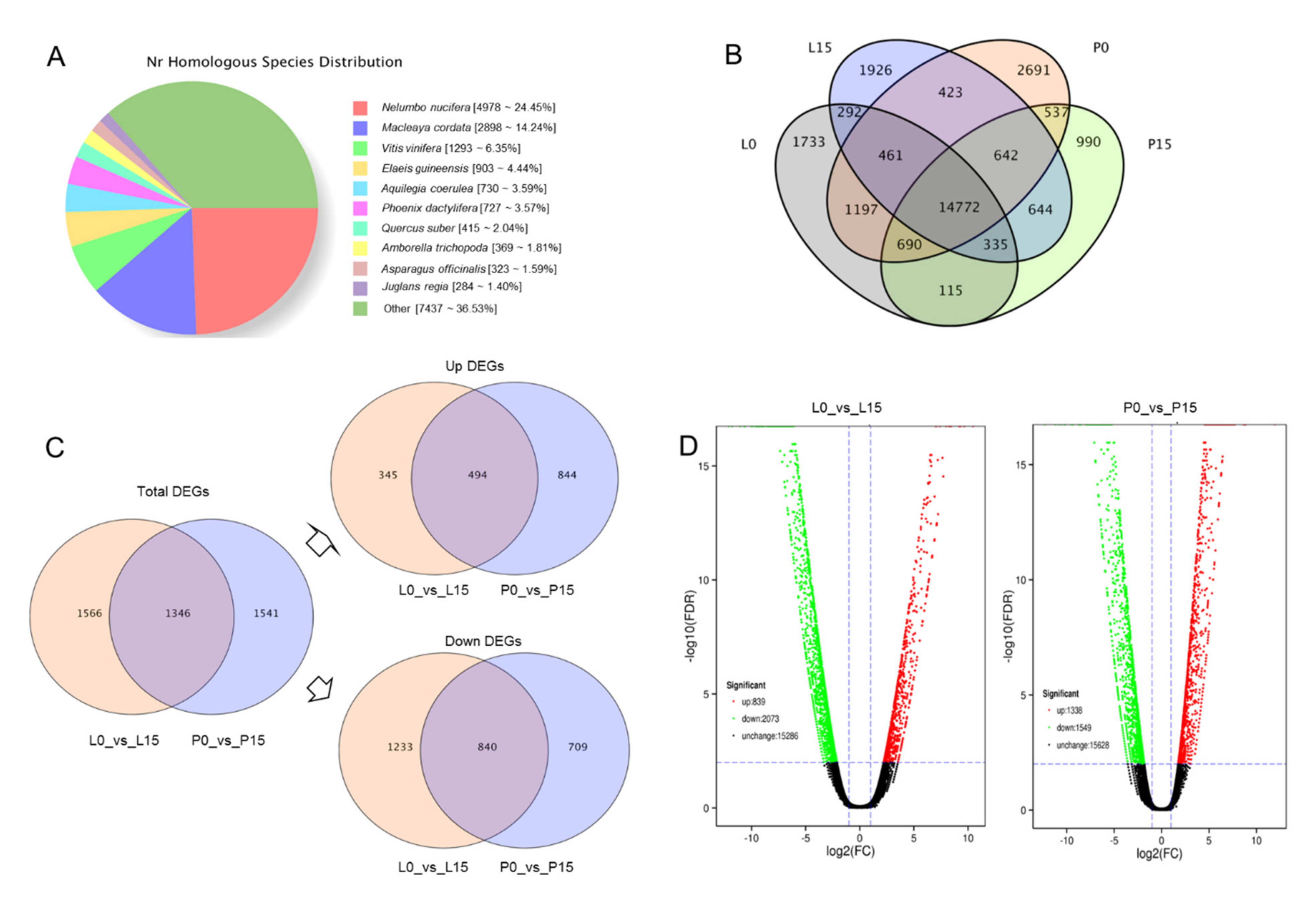
2.3. Comparative Transcriptome Analysis of A. sieboldii under Normal and Drought Treatments
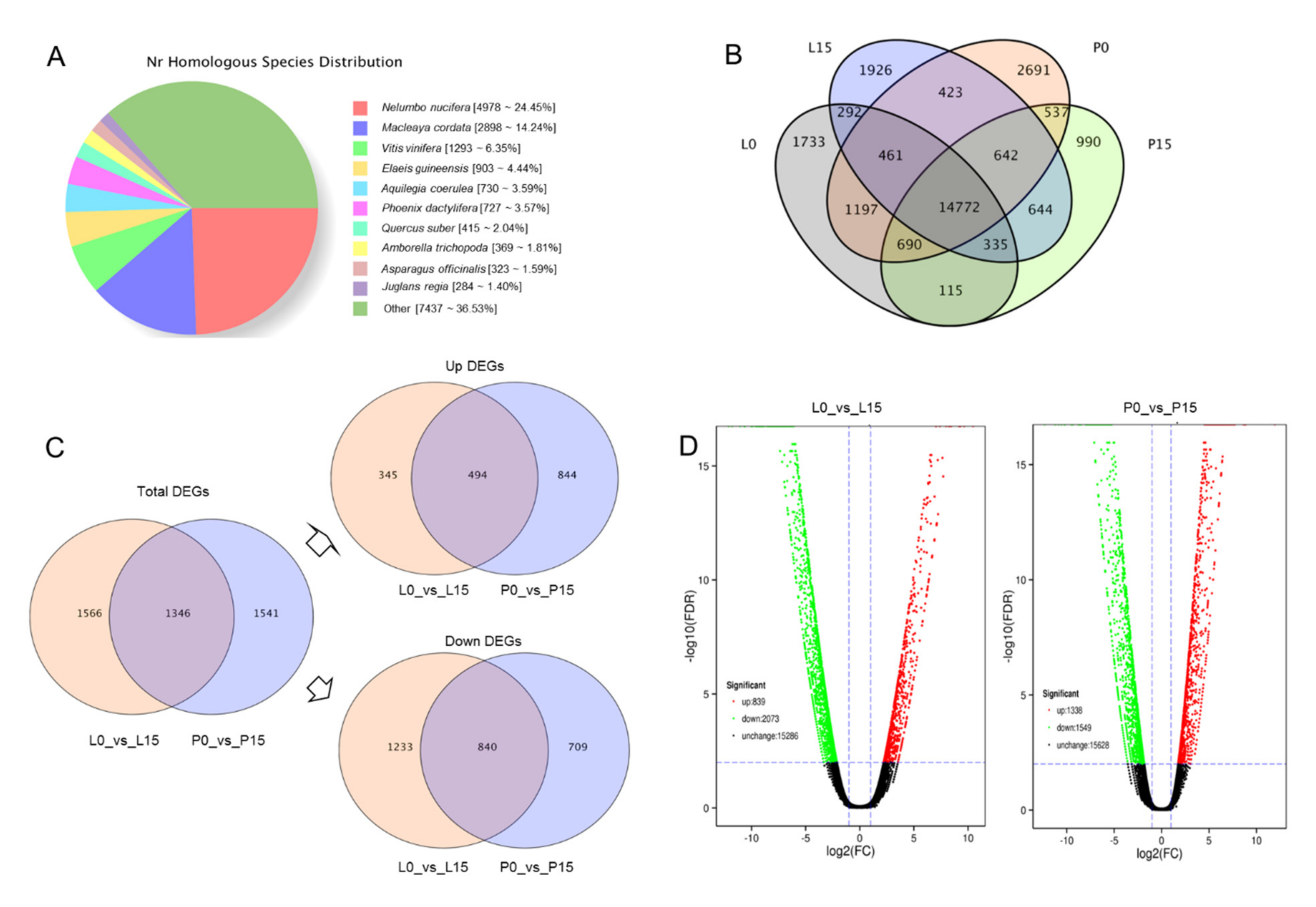
2.3.1. Transcriptome Sequencing and de Novo Assembly
2.3.2. Function Annotation
2.3.3. Analysis of DEGs
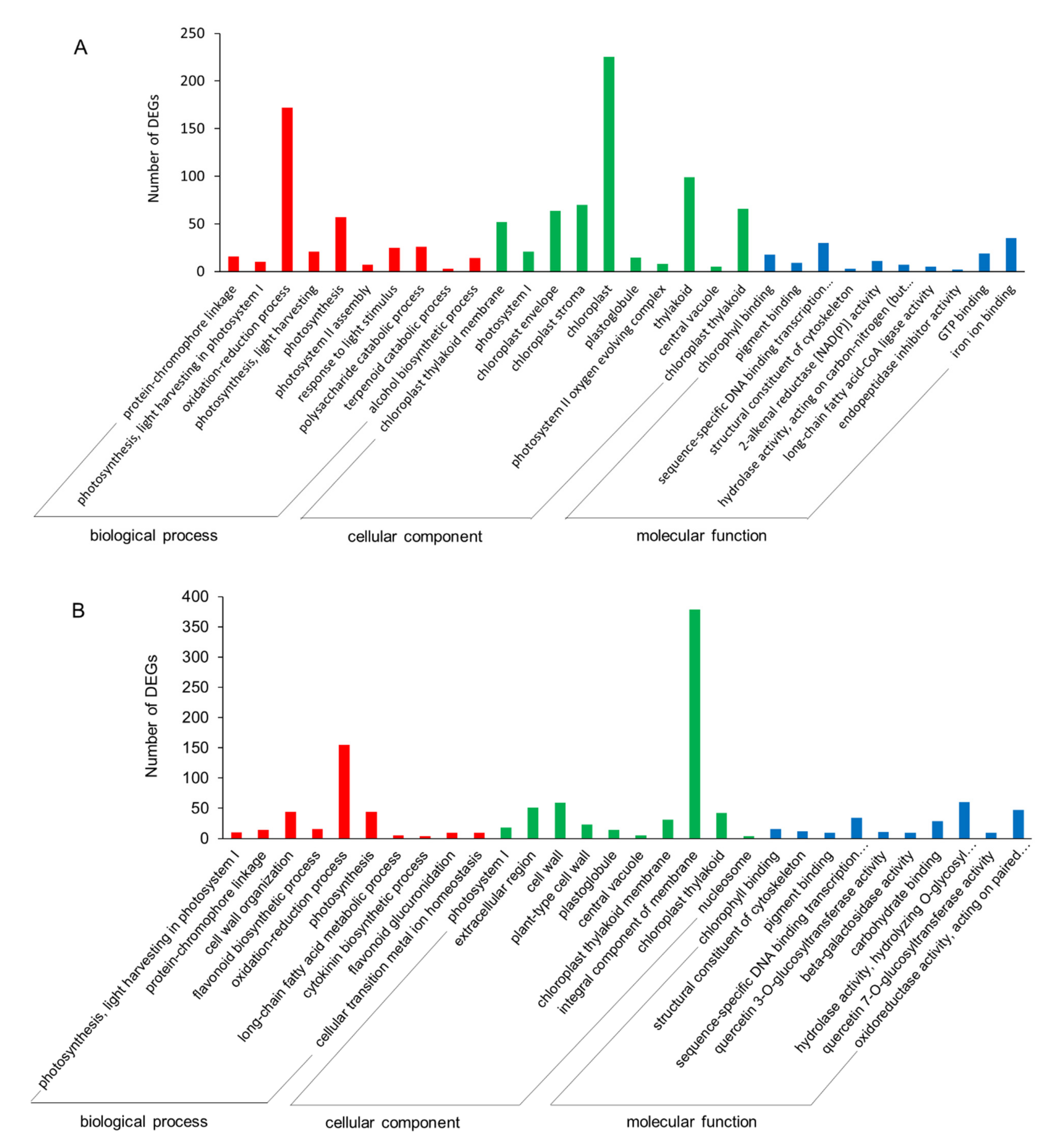
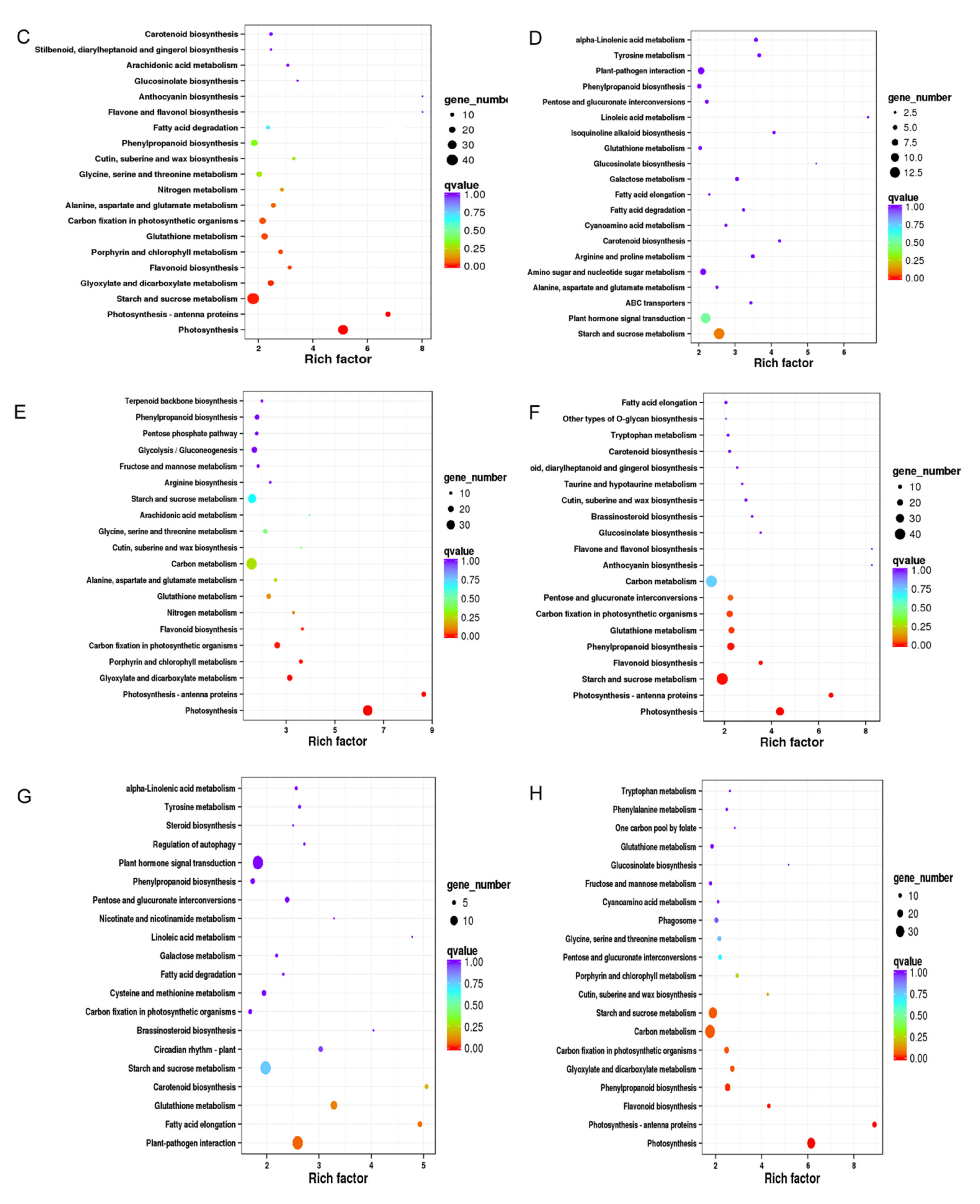
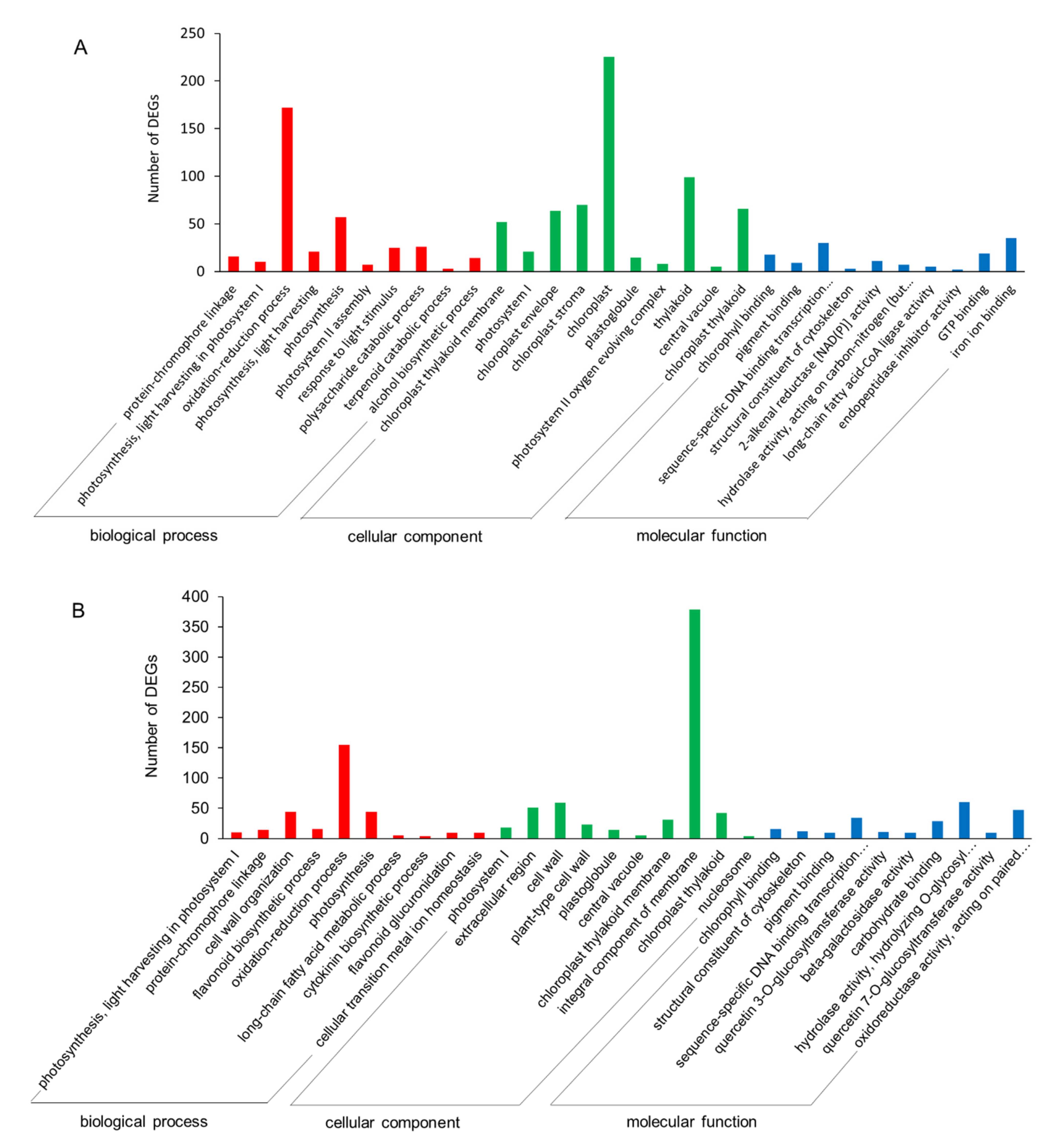
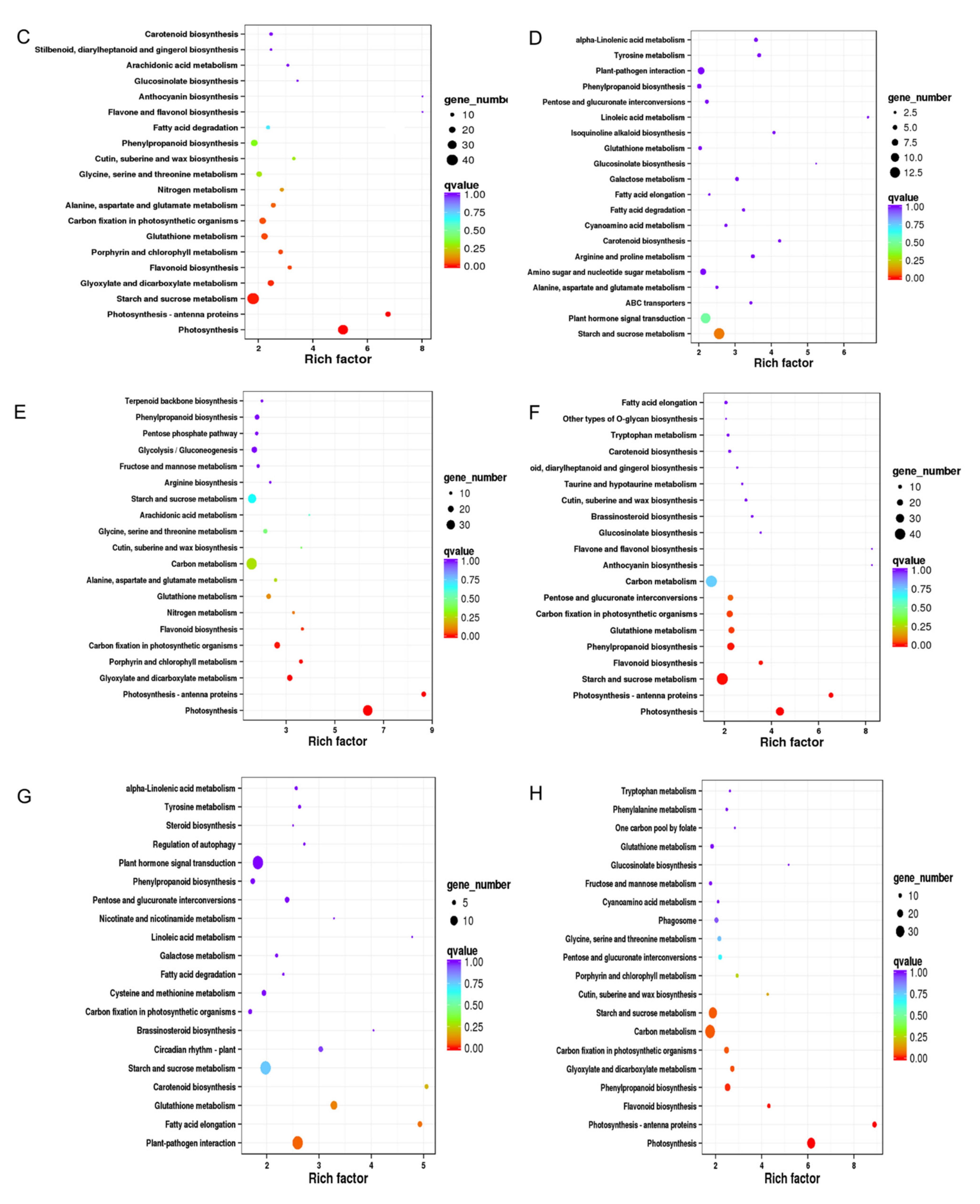
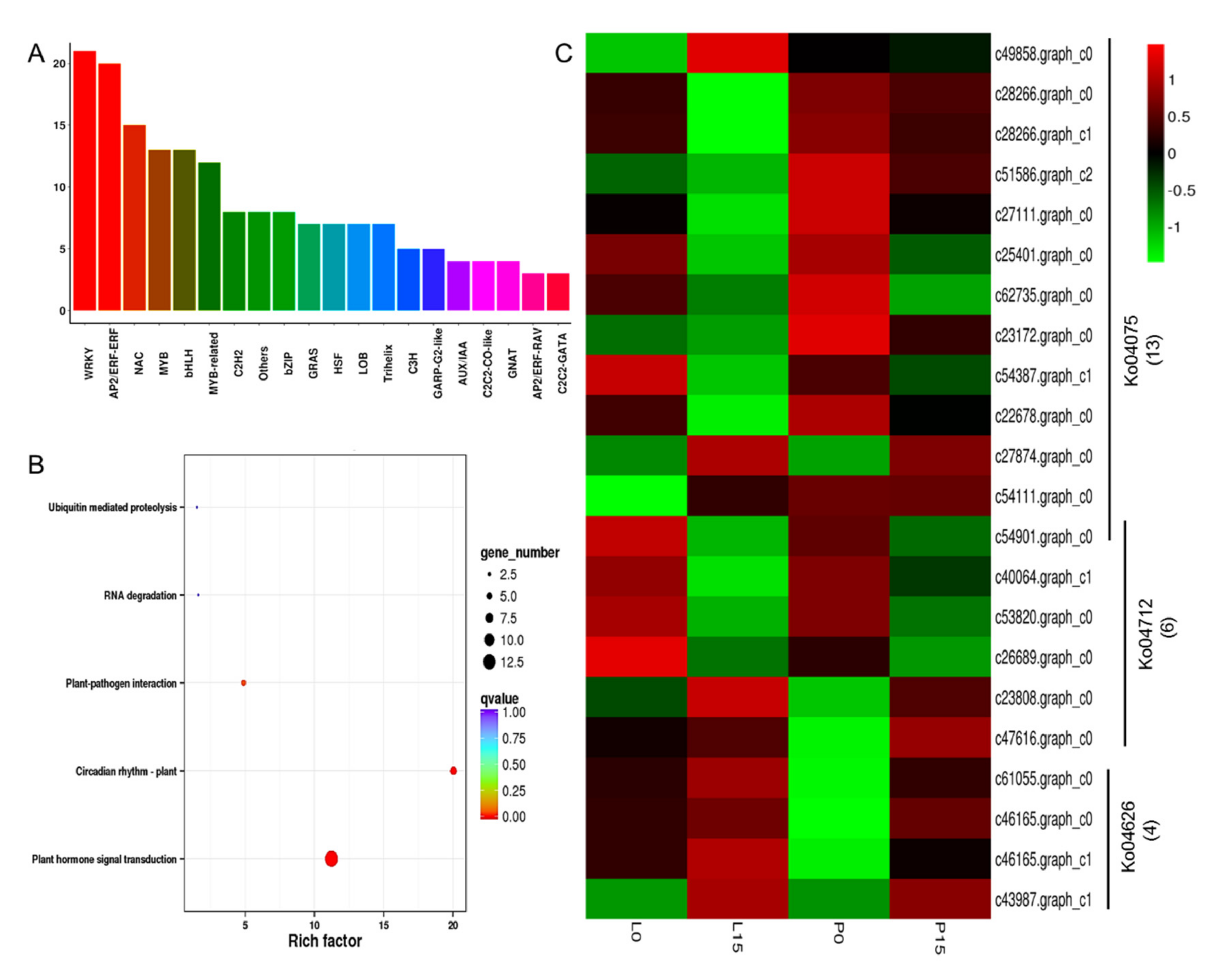
2.3.4. GO and KEGG Enrichments of DEGs
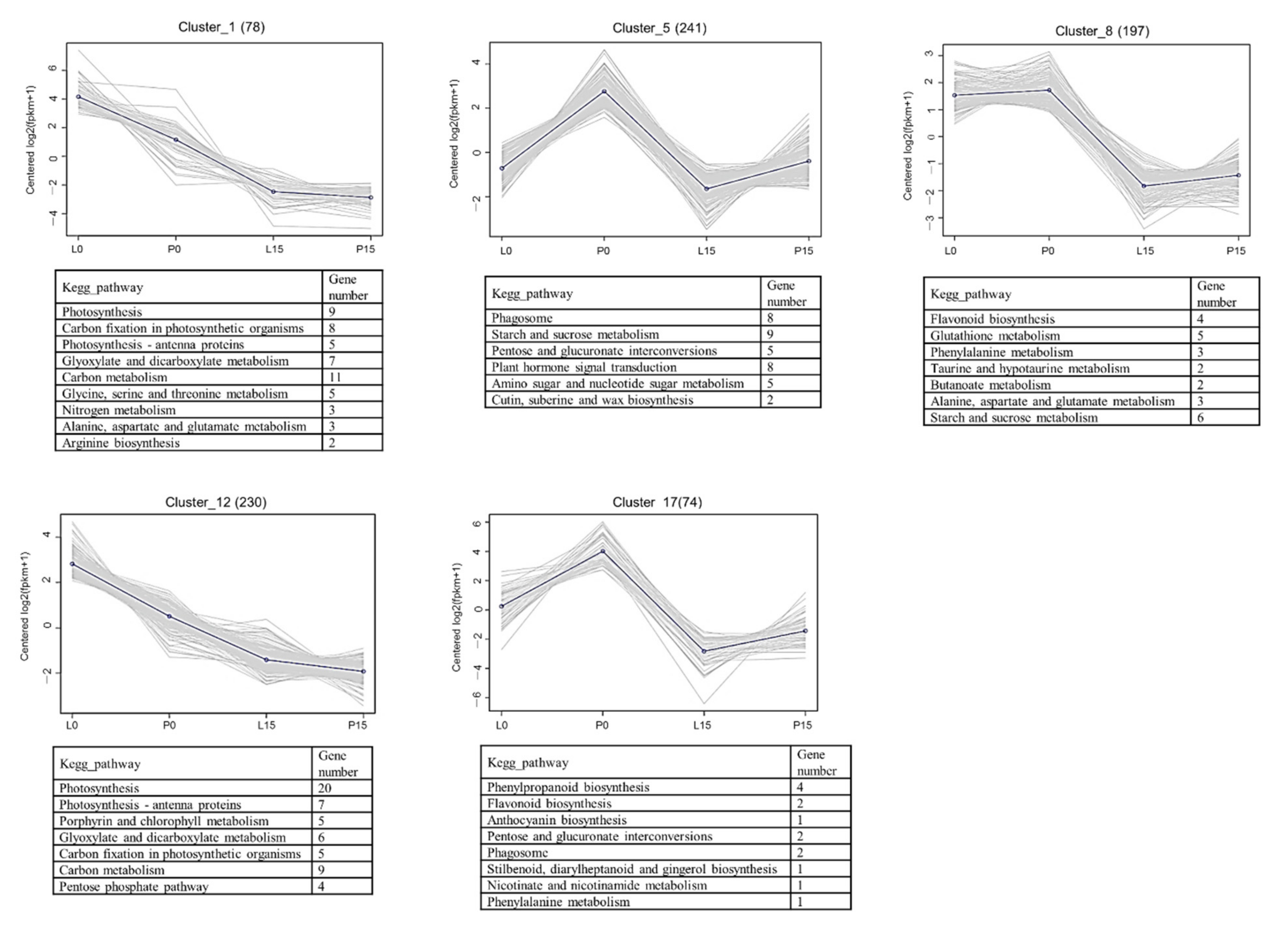
2.3.5. Cluster analysis of DEGs
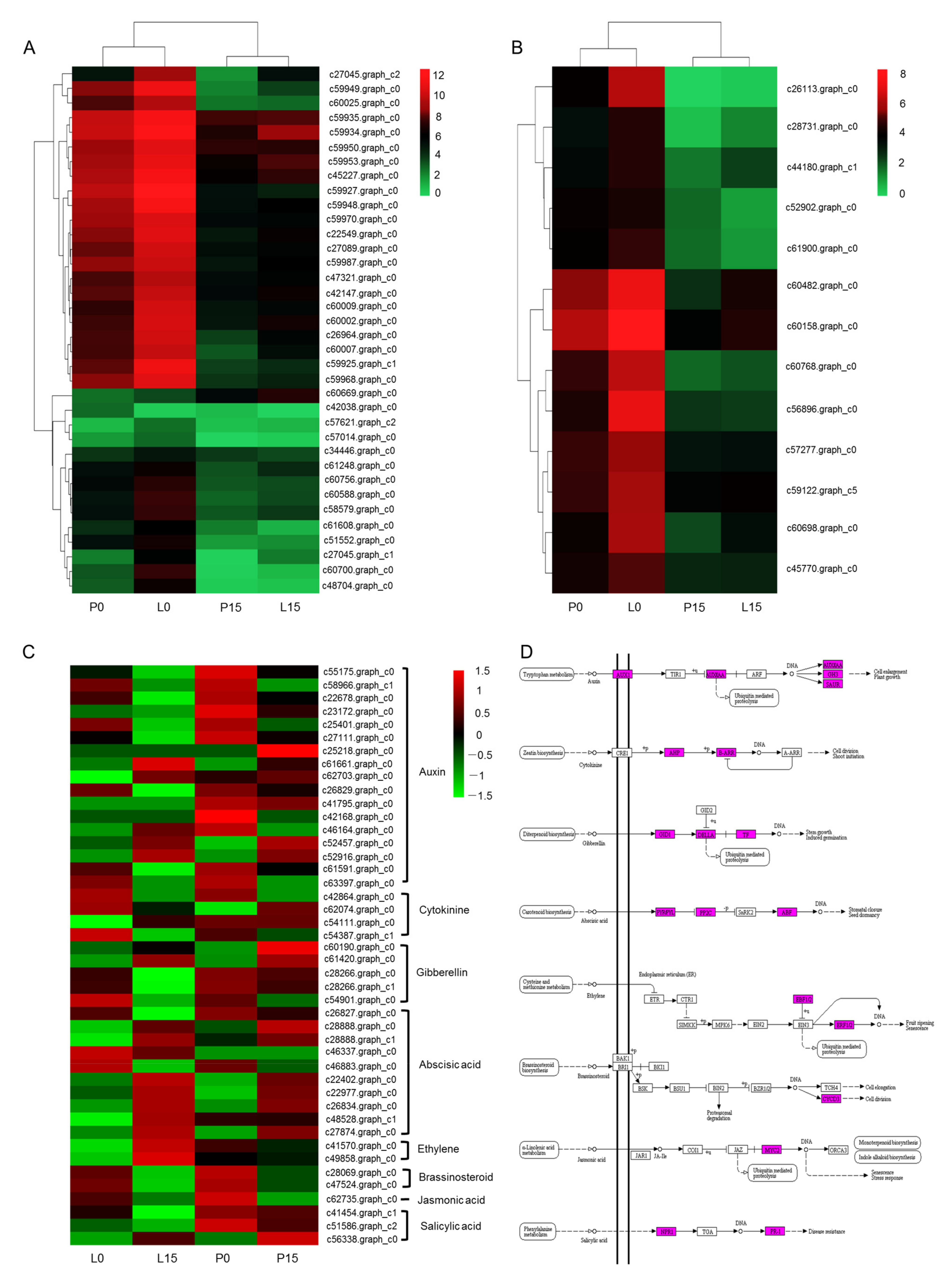
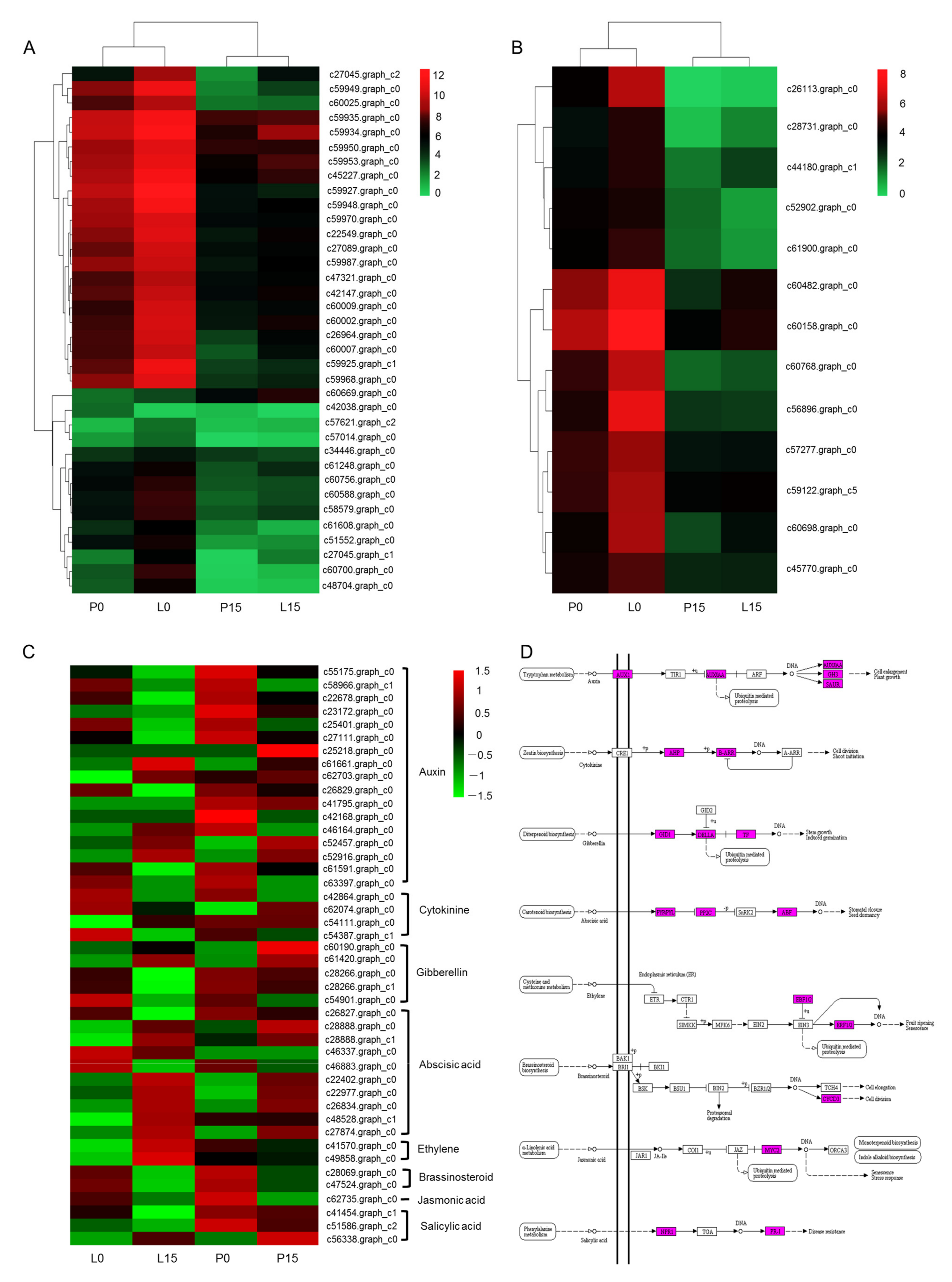
2.3.6. DEGs Related to Photobiosynthesis, Chlorophyll and Hormone Signal Transduction
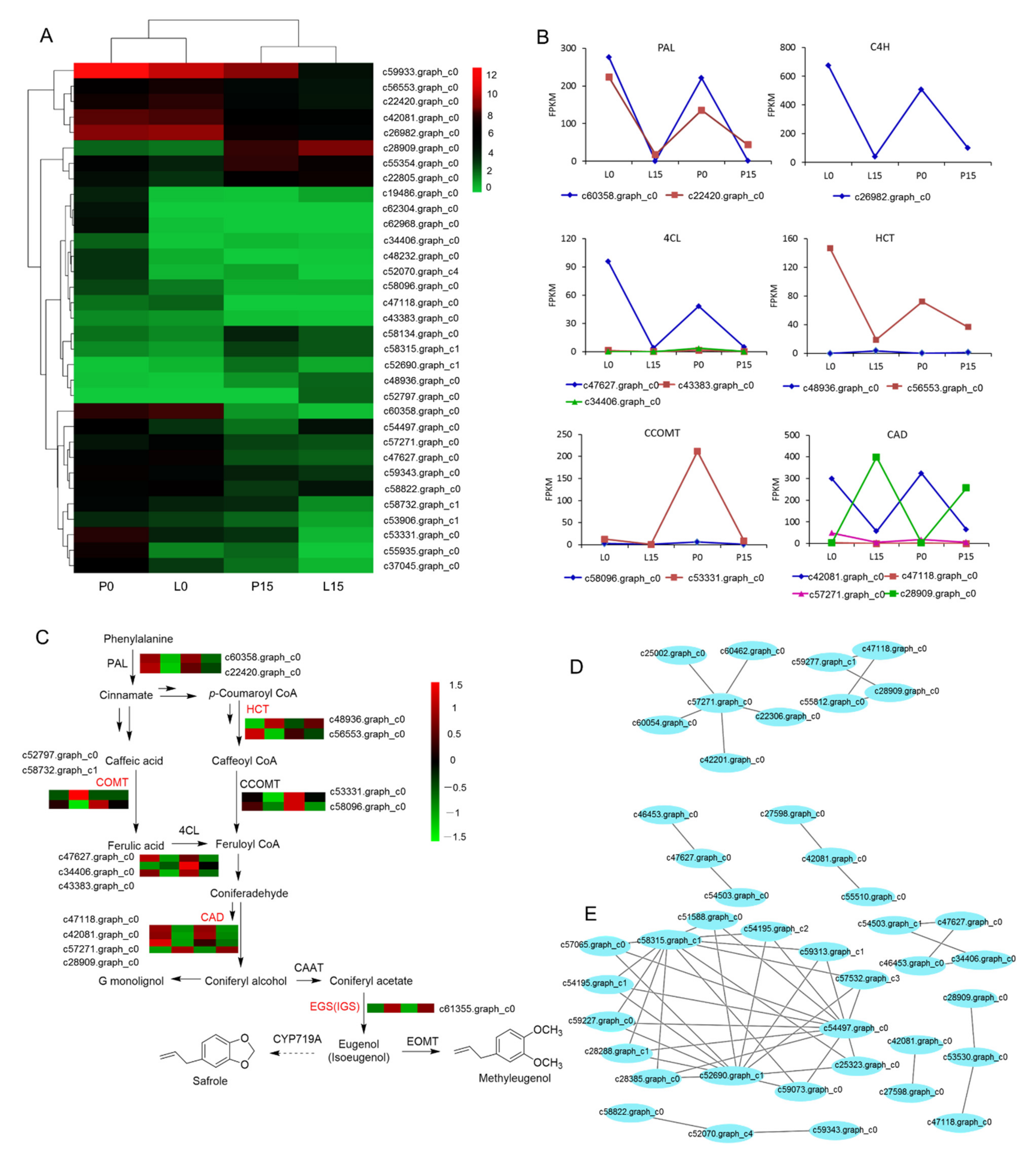
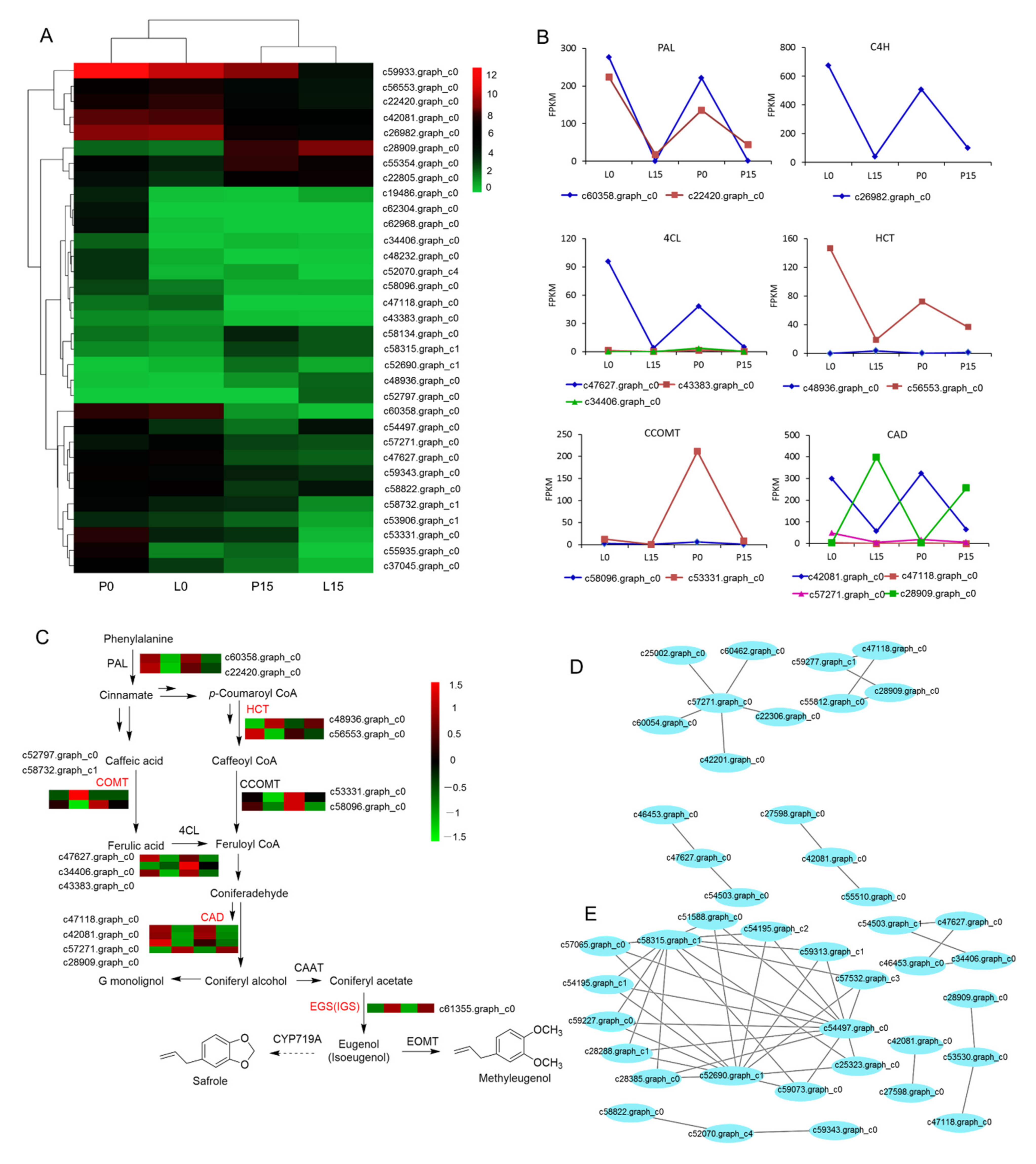
2.3.7. DEGs Associated with Methyleugenol Biobiosynthesis
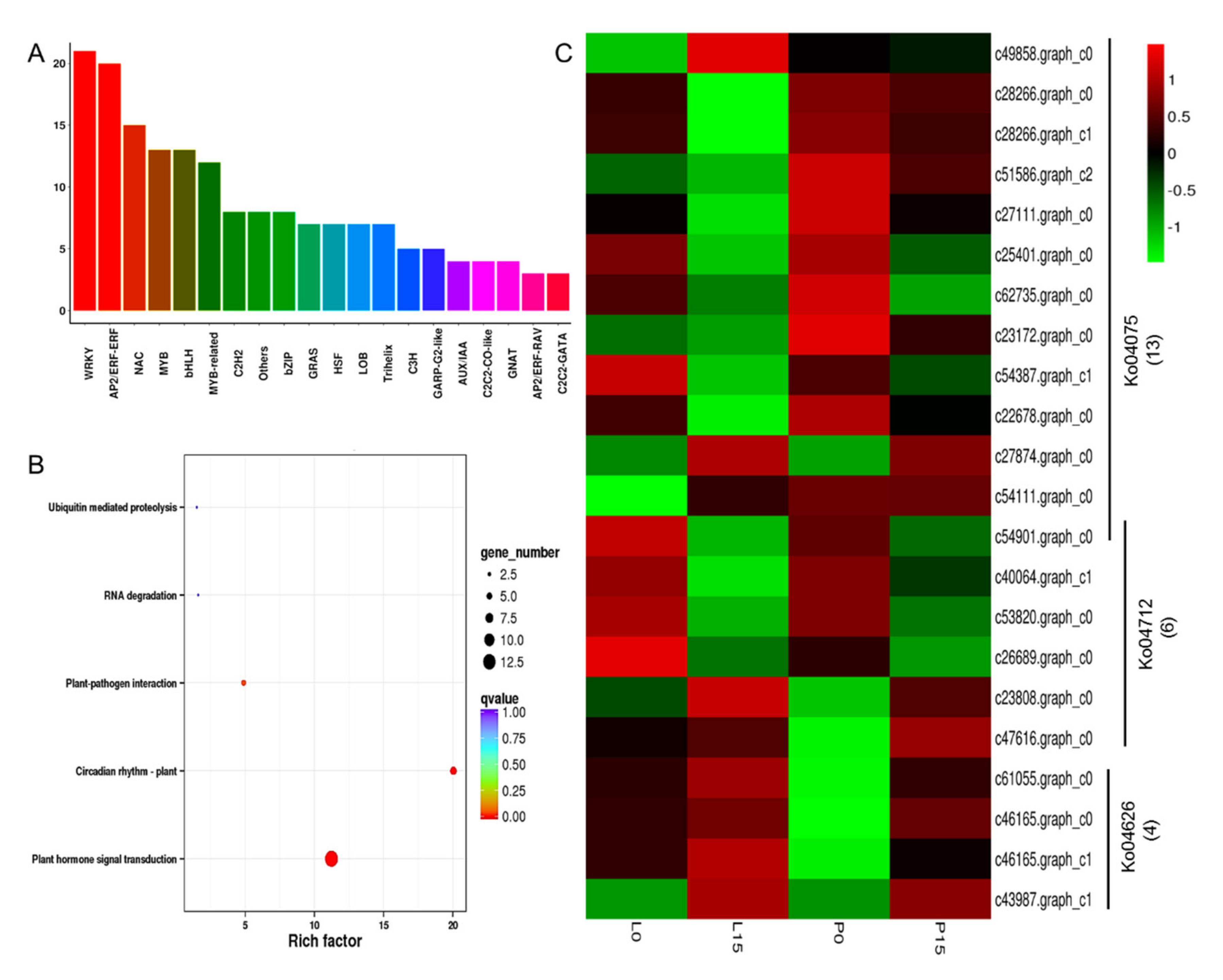
2.3.8. DEGs Encoding Transcription Factors
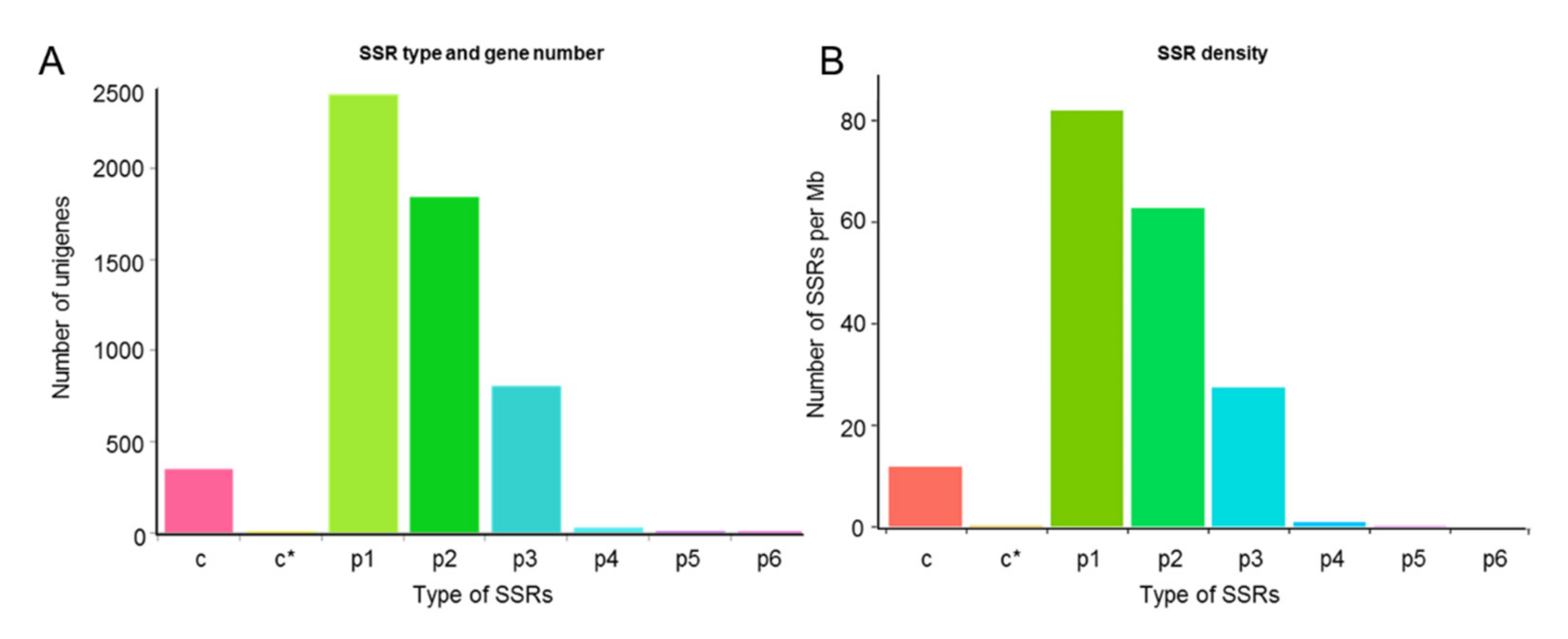
2.3.9. SSR Marker Analysis
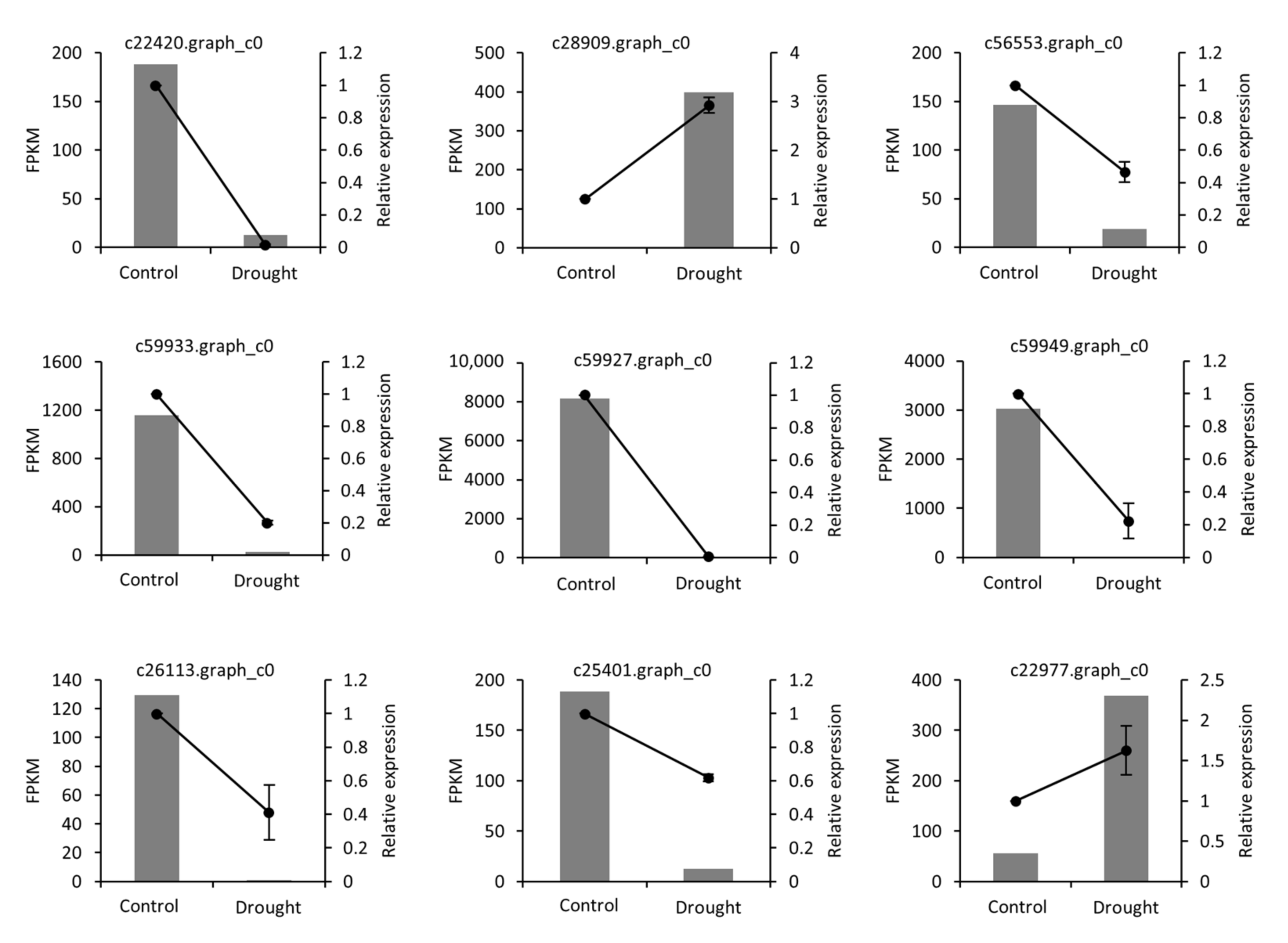
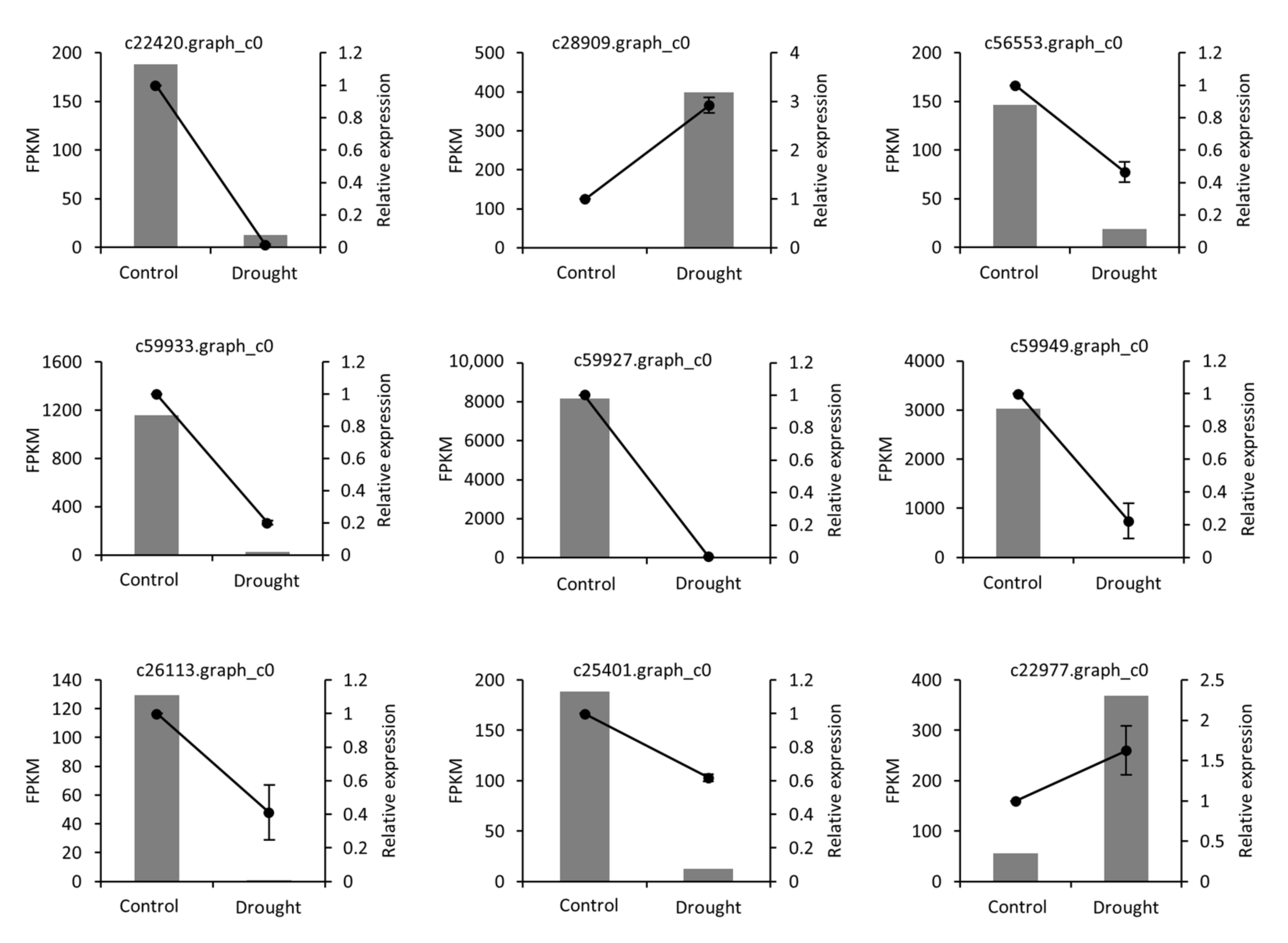
2.4. qRT-PCR Validation
3. Discussion
3.1. Morphological and Physiological Mechanisms in Response to Drought Stress in A. sieboldii
3.2. Effect of Drought Stress on Methyleugenol Biobiosynthesis in Leaf of A. sieboldii
3.3. The Transcription Factors and Plant Hormones Responsive to Drought Stress
3.4. SSR Markers of A. sieboldii Genome
4. Materials and Methods
4.1. Plant Materials and Drought Treatment
4.2. Determination of Chlorophyll and MDA Contents
4.3. Determination of SOD and POD Activities
4.4. GC-MS Analysis of the Major Volatile Bbioactive Constituents
4.5. RNA Extraction, Transcriptome Sequencing, and de Novo Sequence Assembly
4.6. Function Annotation and DEG Analysis
4.7. Validation of Expression Data by qRT-PCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kelly, L. Phylogenetic relationships in Asarum (Aristolochiaceae) based on morphology and ITS sequences. Am. J. Bot. 1998, 85, 1454–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.K. Taxonomy of Asarum Section Asarum (Aristolochiaceae). Syst. Bot. 2001, 26, 17–53. [Google Scholar] [CrossRef]
- Yamaji, H.; Nakamura, T.; Yokoyama, J.; Kondo, K.; Morota, T.; Takeda, S.; Sasaki, H.; Maki, M. A taxonomic study of Asarum sect. Asiasarum (Aristolochiaceae) in Japan. J. Jpn. Bot. 2007, 82, 79–105. [Google Scholar]
- Lee, J.Y.; Moon, S.S.; Hwang, B.K. Isolation and antifungal activity of kakuol, a propiophenone derivative fromAsarum sieboldii rhizome. Pest. Manag. Sci. 2005, 61, 821–825. [Google Scholar] [CrossRef]
- Wu, H.; Li, J.; Zhang, F.; Li, L.; Liu, Z.; He, Z. Essential oil components from Asarum sieboldii Miquel are toxic to the house dust mite Dermatophagoides farinae. Parasitol. Res. 2012, 111, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Spriano, D.; Lehmann, T.; Meier, B. Reduction of Safrole and Methyleugenol in Asari radix et rhizoma by Decoction. Forsch. Komplement. 2009, 16, 162–166. [Google Scholar] [CrossRef]
- Yano, S.; Suzuki, Y.; Yuzurihara, M.; Kase, Y.; Takeda, S.; Watanabe, S.; Aburada, M.; Miyamoto, K.-I. Antinociceptive effect of methyleugenol on formalin-induced hyperalgesia in mice. Eur. J. Pharm. 2006, 553, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ho, S.-H.; Lee, H.-C.; Yap, Y.-L. Insecticidal properties of eugenol, isoeugenol and methyleugenol and their effects on nutrition of Sitophilus zeamais Motsch. (Coleoptera: Curculionidae) and Tribolium castaneum (Herbst) (Coleoptera: Tenebrionidae). J. Stored Prod. Res. 2002, 38, 403–412. [Google Scholar] [CrossRef]
- De Vincenzi, M.; Silano, M.; Stacchini, P.; Scazzocchio, B. Constituents of aromatic plants: I. Methyleugenol. Fitoterapia 2000, 71, 216–221. [Google Scholar] [CrossRef]
- Siano, F.; Ghizzoni, C.; Gionfriddo, F.; Colombo, E.; Servillo, L.; Castaldo, D. Determination of estragole, safrole and eugenol methyl ether in food products. Food Chem. 2003, 81, 469–475. [Google Scholar] [CrossRef]
- Martins, C.; Doran, C.; Laires, A.; Rueff, J.; Rodrigues, A.S. Genotoxic and apoptotic activities of the food flavourings myristicin and eugenol in AA8 and XRCC1 deficient EM9 cells. Food Chem. Toxicol. 2011, 49, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Wang, Z. Protection of idioplasm resources of Asarum sieboldii Miq. J. Shaanxi Norm. Univ. 2006, 34, 81–84. [Google Scholar]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gao, X.; Li, J.; Gong, X.; Yang, P.; Gao, J.; Wang, P.; Feng, B. Comparative analysis of proso millet (Panicum miliaceum L.) leaf transcriptomes for insight into drought tolerance mechanisms. BMC Plant Biol. 2019, 19, 397. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Ullah, F.; Ben-Hur, A.; Reddy, A.S.N. Transcriptome Analysis of Drought-Resistant and Drought-Sensitive Sorghum (Sorghum bicolor) Genotypes in Response to PEG-Induced Drought Stress. Int. J. Mol. Sci. 2020, 21, 772–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Deng, J.L.; Tian, Y. Transcriptome sequencing of the apricot (Prunus armeniaca L.) and identification of differentially expressed genes involved in drought stress. Phytochemistry 2020, 171, 112226. [Google Scholar] [CrossRef]
- Miyashita, K.; Tanakamaru, S.; Maitani, T.; Kimura, K. Recovery responses of photosynthesis, transpiration, and stomatal conductance in kidney bean following drought stress. Env. Exp. Bot. 2005, 53, 205–214. [Google Scholar] [CrossRef]
- Pichersky, E.; Noel, J.P.; Dudareva, N. Biosynthesis of Plant Volatiles: Nature’s Diversity and Ingenuity. Science 2006, 311, 808–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, Y.; Davidovich-Rikanati, R.; Fridman, E.; Gang, D.R.; Bar, E.; Lewinsohn, E.; Pichersky, E. The Biochemical and Molecular Basis for the Divergent Patterns in the Biosynthesis of Terpenes and Phenylpropenes in the Peltate Glands of Three Cultivars of Basil. Plant Physiol. 2004, 136, 3724–3736. [Google Scholar] [CrossRef] [Green Version]
- Mandoulakani, B.A.; Eyvazpour, E.; Ghadimzadeh, M. The effect of drought stress on the expression of key genes involved in the biosynthesis of phenylpropanoids and essential oil components in basil (Ocimum basilicum L.). Phytochemistry 2017, 139, 1–7. [Google Scholar] [CrossRef]
- Nasrollahi, V.; Mirzaie-asl, A.; Piri, K.; Nazeri, S.; Mehrabi, R. The effect of drought stress on the expression of key genes involved in the biobiosynthesis of triterpenoid saponins in liquorice (Glycyrrhiza glabra). Phytochemistry 2014, 103, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Said-Al Ahl, H.A.H.; Omer, E.A.; Naguib, N.Y. Effect of water stress and nitrogen fertilizer on herb and essential oil of ore-gano. Int. Agrophys. 2009, 23, 269–275. [Google Scholar]
- Sánchez-Blanco, M.J.; Álvarez, S.; Navarro, A.; Bañón, S. Changes in leaf water relations, gas exchange, growth and flowering quality in potted geranium plants irrigated with different water regimes. J. Plant. Physiol. 2009, 166, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, S.; Bennett, L.T. Photosynthesis, photochemistry and antioxidative defence in response to two drought severities and with re-watering inAllocasuarina luehmannii. Plant Biol. 2009, 11, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ma, X.; Guo, H.; Sukiran, N.L.; Guo, B.; Assmann, S.M.; Ma, H. Flower Development under Drought Stress: Morphological and Transcriptomic Analyses Reveal Acute Responses and Long-Term Acclimation in Arabidopsis. Plant Cell 2013, 25, 3785–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Ali, T.; Liu, Z. Comparative Transcriptomic Analysis Reveals the Effects of Drought on the Biosynthesis of Methyleugenol in Asarum sieboldii Miq. Biomolecules 2021, 11, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Monneveux, P.; Ramirez, D.; Pino, M.-T. Drought tolerance in potato (S. tuberosum L.). Plant Sci. 2013, 205-206, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Vanderbeld, B.; Wan, J.; Huang, Y. Narrowing Down the Targets: Towards Successful Genetic Engineering of Drought-Tolerant Crops. Mol. Plant 2010, 3, 469–490. [Google Scholar] [CrossRef]
- De Block, M.; Verduyn, C.; De Brouwer, D.; Cornelissen, M. Poly(ADP-ribose) polymerase in plants affects energy homeostasis, cell death and stress tolerance. Plant J. 2004, 41, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shiroto, Y.; Kishitani, S.; Ito, Y.; Toriyama, K. Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 2008, 28, 21–30. [Google Scholar] [CrossRef]
- Pellegrineschi, A.; Reynolds, M.P.; Pacheco, M.; Brito, R.M.; Almeraya, R.; Yamaguchi-Shinozaki, K.; Hoisington, D. Stress-induced expression in wheat of the Arabidopsis thaliana DREB1A gene delays water stress symptoms under greenhouse conditions. Genome 2004, 47, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-R.; Yang, A.-F.; Yue, G.-D.; Gao, Q.; Yin, H.-Y.; Zhang, J.-R. Enhanced expression of phospholipase C 1 (ZmPLC1) improves drought tolerance in transgenic maize. Planta 2008, 227, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.A.S.; Faria, J.A.Q.A.; Soares-Ramos, J.R.L.; Reis, P.A.B.; Pinheiro, G.L.; Piovesan, N.D.; Morais, A.T.; Menezes, C.C.; Cano, M.A.O.; Fietto, L.G.; et al. The ER luminal binding protein (BiP) mediates an increase in drought tolerance in soybean and delays drought-induced leaf senescence in soybean and tobacco. J. Exp. Bot. 2009, 60, 533–546. [Google Scholar] [CrossRef]
- Zhao, X.; Li, C.; Wan, S.; Zhang, T.; Yan, C.; Shan, S. Transcriptomic analysis and discovery of genes in the response of Arachis hypogaea to drought stress. Mol. Biol. Rep. 2018, 45, 119–131. [Google Scholar] [CrossRef]
- Ye, G.; Ma, Y.; Feng, Z.; Zhang, X. Transcriptomic analysis of drought stress responses of sea buckthorn (Hippophae rhamnoide ssp. sinensis) by RNA-Seq. PLoS ONE 2018, 13, e0202213. [Google Scholar] [CrossRef] [Green Version]
- Heller, S. Computer Software Reviews. NIST Structures and Properties Database and Estimation Program. J. Chem. Inf. Comput. Sci. 1991, 31, 432–434. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Xiong, L. General mechanisms of drought response and their application in drought resistance improvement in plants. Cell. Mol. Life Sci. 2015, 72, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, T.C.; O’Toole, J.C.; Yambao, E.B.; Turner, N.C. Influence of Osmotic Adjustment on Leaf Rolling and Tissue Death in Rice (Oryza sativa L.). Plant Physiol. 1984, 75, 338–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.-H.; Guo, P.-G.; Michael, B.; Stefania, G.; Salvatore, C. Evaluation of Chlorophyll Content and Fluorescence Parameters as Indicators of Drought Tolerance in Barley. Agric. Sci. China 2006, 5, 751–757. [Google Scholar] [CrossRef]
- Pinheiro, C.; Chaves, M.M. Photobiosynthesis and drought: Can we make metabolic connections from available data? J. Exp. Bot. 2011, 62, 869–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Liu, M.; Gu, W.; Chen, Z.; Gu, Y.; Pei, L.; Tian, R. Effect of drought on photosynthesis, total antioxidant capacity, bioactive component accumulation, and the transcriptome of Atractylodes lancea. BMC Plant Biol. 2021, 21, 293. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yao, Q.; Patil, G.; Agarwal, G.; Deshmukh, R.; Lin, L.; Wang, B.; Wang, Y.; Prince, S.J.; Song, L.; et al. Identification and Comparative Analysis of Differential Gene Expression in Soybean Leaf Tissue under Drought and Flooding Stress Revealed by RNA-Seq. Front. Plant Sci. 2016, 7, 1044. [Google Scholar] [CrossRef] [Green Version]
- Tabaeizadeh, Z. Drought-Induced Responses in Plant Cells. Int. Rev. Cytol. 1998, 182, 193–247. [Google Scholar] [CrossRef]
- Shao, H.-B.; Chu, L.-Y.; Lu, Z.-H.; Kang, C.-M. Primary antioxidant free radical scavenging and redox signaling pathways in higher plant cells. Int. J. Biol. Sci. 2008, 4, 8–14. [Google Scholar] [CrossRef]
- Pandey, H.C.; Baig, M.J.; Chandra, A.; Bhatt, R.K. Drought stress induced changes in lipid peroxidation and antioxidant system in genus Avena. J. Environ. Biol. 2010, 31, 435–440. [Google Scholar] [PubMed]
- Maraghni, M.; Gorai, M.; Neffati, M.; Van Labeke, M.C. Differential responses to drought stress in leaves and roots of wild jujube, Ziziphus lotus. Acta Physiol. Plant 2014, 36, 945–953. [Google Scholar] [CrossRef]
- Fayez, K.A.; Bazaid, S.A. Improving drought and salinity tolerance in barley by application of salicylic acid and potassium nitrate. J. Saudi Soc. Agric. Sci. 2014, 13, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Vardharajula, S.; Skz, A.; Grover, M.; Reddy, G.; Venkateswarlu, B. Drought-tolerant plant growth promoting Bacillus spp.: Effect on growth, osmolytes, and antioxidant status of maize under drought stress. J. Plant Interact. 2011, 6, 1–14. [Google Scholar]
- Wu, X.; Zhu, Z.; Li, X.; Zha, D. Effects of cytokinin on photosynthetic gas exchange, chlorophyll fluorescence parameters and antioxidative system in seedlings of eggplant (Solanum melongena L.) under salinity stress. Acta Physiol. Plant 2012, 34, 2105–2114. [Google Scholar] [CrossRef]
- Simon, J.E.; Reiss-Bubenheim, D.; Joly, R.J.; Charles, D.J. Water Stress-Induced Alterations in Essential Oil Content and Composition of Sweet Basil. J. Essent. Oil Res. 1992, 4, 71–75. [Google Scholar] [CrossRef]
- Fatima, S.; Farooqi, A.H.A.; Ansari, S.R.; Sharma, S. Effect of Water Stress on Growth and Essential Oil Metabolism inCymbopogon martinii(Palmarosa) Cultivars. J. Essent. Oil Res. 1999, 11, 491–496. [Google Scholar] [CrossRef]
- Burbott, A.J.; Loomis, W.D. Evidence for Metabolic Turnover of Monoterpenes in Peppermint. Plant Physiol. 1969, 44, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Koeduka, T.; Louie, G.V.; Orlova, I.; Kish, C.M.; Ibdah, M.; Wilkerson, C.G.; Bowman, M.E.; Baiga, T.J.; Noel, J.P.; Dudareva, N.; et al. The multiple phenylpropene synthases in both Clarkia breweri and Petunia hybrida represent two distinct protein lineages. Plant J. 2008, 54, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, C.; Zhang, H.; Liu, F.; Ma, D.; Liu, Z. Comparative Transcriptomics Analysis for Gene Mining and Identification of a Cinnamyl Alcohol Dehydrogenase Involved in Methyleugenol Biosynthesis from Asarum sieboldii Miq. Molecules 2018, 23, 3184. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [Green Version]
- Gahlaut, V.; Jaiswal, V.; Kumar, A.; Gupta, P.K. Transcription factors involved in drought tolerance and their possible role in developing drought tolerant cultivars with emphasis on wheat (Triticum aestivum L.). Appl. Genet. 2016, 129, 2019–2042. [Google Scholar] [CrossRef] [PubMed]
- Golldack, D.; Lüking, I.; Yang, O. Plant tolerance to drought and salinity: Stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 2011, 30, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nolan, T.; Jiang, H.; Yin, Y. AP2/ERF Transcription Factor Regulatory Networks in Hormone and Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2019, 10, 228–245. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1026. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.-Y.; Du, Y.-T.; Ma, J.; Min, D.-H.; Jin, L.-G.; Chen, J.; Chen, M.; Zhou, Y.-B.; Ma, Y.-Z.; Xu, Z.-S.; et al. The WRKY Transcription Factor GmWRKY12 Confers Drought and Salt Tolerance in Soybean. Int. J. Mol. Sci. 2018, 19, 4087. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Chen, M.; Li, L.; Xu, Z.; Chen, X.; Guo, J.; Ma, Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J. Exp. Bot. 2009, 60, 3781–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.L.; Zhang, C.L.; Wang, G.L.; Wang, Y.X.; Qi, C.H.; You, C.X.; Li, Y.Y.; Hao, Y.J. Apple AP2/EREBP transcription factor MdSHINE2 confers drought resistance by regulating wax biosynthesis. Planta 2019, 249, 1627–1643. [Google Scholar] [CrossRef]
- Campalans, A.; Messeguer, R.; Goday, A.; Pagès, M. Plant responses to drought, from ABA signal transduction events to the action of the induced proteins. Plant Physiol. Biochem. 1999, 37, 327–340. [Google Scholar] [CrossRef]
- Huang, D.; Wu, W.; Abrams, S.R.; Cutler, A.J. The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J. Exp. Bot. 2008, 59, 2991–3007. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.; Wani, S.; Singh, B.; Bohra, A.; Dar, Z.; Lone, A.; Pareek, A.; Singla-Pareek, S.L. Transcription Factors and Plants Response to Drought Stress: Current Understanding and Future Directions. Front. Plant Sci. 2016, 7, 1029–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flexas, J.; Medrano, H. Drought-inhibition of photobiosynthesis in C3 plants: Stomatal and non-stomatal limitations revisited. Ann. Bot. 2002, 89, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.; Davies, W.J. ABA-based chemical signalling: The co-ordination of responses to stress in plants. Plant Cell Env. 2002, 25, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Gosti, F.; Beaudoin, N.; Serizet, C.; Webb, A.A.R.; Vartanian, N.; Giraudat, J. ABI1 Protein Phosphatase 2C Is a Negative Regulator of Abscisic Acid Signaling. Plant Cell 1999, 11, 1897–1910. [Google Scholar] [CrossRef] [Green Version]
- Paponov, I.A.; Paponov, M.; Teale, W.; Menges, M.; Chakrabortee, S.; Murray, J.A.; Palme, K. Comprehensive Transcriptome Analysis of Auxin Responses in Arabidopsis. Mol. Plant 2008, 1, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmasov, T.; Murfett, J.; Hagen, G.; Guilfoyle, T.J. Aux/IAA proteins repress expression of reporter genes containing natural and highly active synthetic auxin response elements. Plant Cell 1997, 9, 1963–1971. [Google Scholar]
- Tiwari, S.B.; Wang, X.J.; Hagen, G.; Guilfoyle, T.J. AUX/IAA proteins are active repressors, and their stability and activity are modulated by auxin. Plant Cell 2001, 13, 2809–2822. [Google Scholar] [CrossRef] [Green Version]
- Ulmasov, T.; Hagen, G.; Guilfoyle, T.J. Activation and repression of transcription by auxin-response factors. Proc. Natl. Acad. Sci. USA 1999, 96, 5844–5849. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, X.; Zhang, D.; Tang, H.; Sun, B.; Li, C.; Hao, L.; Liu, C.; Li, Y.; Shi, Y.; et al. Genome-wide identification of gene expression in contrasting maize inbred lines under field drought conditions reveals the significance of transcription factors in drought tolerance. PLoS ONE 2017, 12, e0179477. [Google Scholar] [CrossRef] [Green Version]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Zhu, H.; Zhou, Y.Y.; Zhai, H.; He, S.Z.; Zhao, N.; Liu, Q.C. Transcriptome profiling reveals insights into the molecular mecha-nism of drought tolerance in sweetpotato. J. Integr. Agric. 2019, 18, 9–23. [Google Scholar] [CrossRef]
- Durand, J.; Bodénès, C.; Chancerel, E.; Frigerio, J.-M.; Vendramin, G.; Sebastiani, F.; Buonamici, A.; Gailing, O.; Koelewijn, H.-P.; Villani, F.; et al. A fast and cost-effective approach to develop and map EST-SSR markers: Oak as a case study. BMC Genom. 2010, 11, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Yang, C.; Tian, B.; Yang, J.-B.; Liu, A. Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communisL.). BMC Plant Biol. 2010, 10, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, L.P. Spectrophotometric Determination of Chlorophylls and Pheophytins in Plant Extracts-Corrections. Anal. Chem. 1960, 32, 1414. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its locali-zation. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Tatusov, R.L.; Galperin, M.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, S.K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in Data: An Introduction to Cluster Analysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range | Transcript | Unigene |
|---|---|---|
| 300–500 | 33,009 (21.91%) | 18,524 (41.15%) |
| 500–1000 | 36,657 (24.33%) | 12,228 (27.16%) |
| 1000–2000 | 43,693 (29.00%) | 8635 (19.18%) |
| 2000+ | 37,316 (24.77%) | 5629 (12.50%) |
| Total Number | 150,675 | 45,016 |
| Total Length | 218,185,217 | 44,921,544 |
| N50 Length | 2097 | 1545 |
| Database | Annotated Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| COG | 6226 | 1303 | 4923 |
| GO | 11,184 | 4030 | 7154 |
| KEGG | 7574 | 2610 | 4964 |
| KOG | 12,185 | 4455 | 7730 |
| Pfam | 14,251 | 3973 | 10,278 |
| Swissprot | 14,415 | 4988 | 9427 |
| eggNOG | 19,257 | 7278 | 11,979 |
| Nr | 20,366 | 8170 | 12,196 |
| All_Annotated | 20,551 | 8324 | 12,227 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Ali, T.; Liu, Z. Physiological, Biochemical and Transcriptomic Analysis of the Aerial Parts (Leaf-Blade and Petiole) of Asarum sieboldii Responding to Drought Stress. Int. J. Mol. Sci. 2021, 22, 13402. https://doi.org/10.3390/ijms222413402
Liu F, Ali T, Liu Z. Physiological, Biochemical and Transcriptomic Analysis of the Aerial Parts (Leaf-Blade and Petiole) of Asarum sieboldii Responding to Drought Stress. International Journal of Molecular Sciences. 2021; 22(24):13402. https://doi.org/10.3390/ijms222413402
Chicago/Turabian StyleLiu, Fawang, Tahir Ali, and Zhong Liu. 2021. "Physiological, Biochemical and Transcriptomic Analysis of the Aerial Parts (Leaf-Blade and Petiole) of Asarum sieboldii Responding to Drought Stress" International Journal of Molecular Sciences 22, no. 24: 13402. https://doi.org/10.3390/ijms222413402
APA StyleLiu, F., Ali, T., & Liu, Z. (2021). Physiological, Biochemical and Transcriptomic Analysis of the Aerial Parts (Leaf-Blade and Petiole) of Asarum sieboldii Responding to Drought Stress. International Journal of Molecular Sciences, 22(24), 13402. https://doi.org/10.3390/ijms222413402