
MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MicroRNA-16 Is Downregulated in NSCLC Primary Samples and Cell Lines
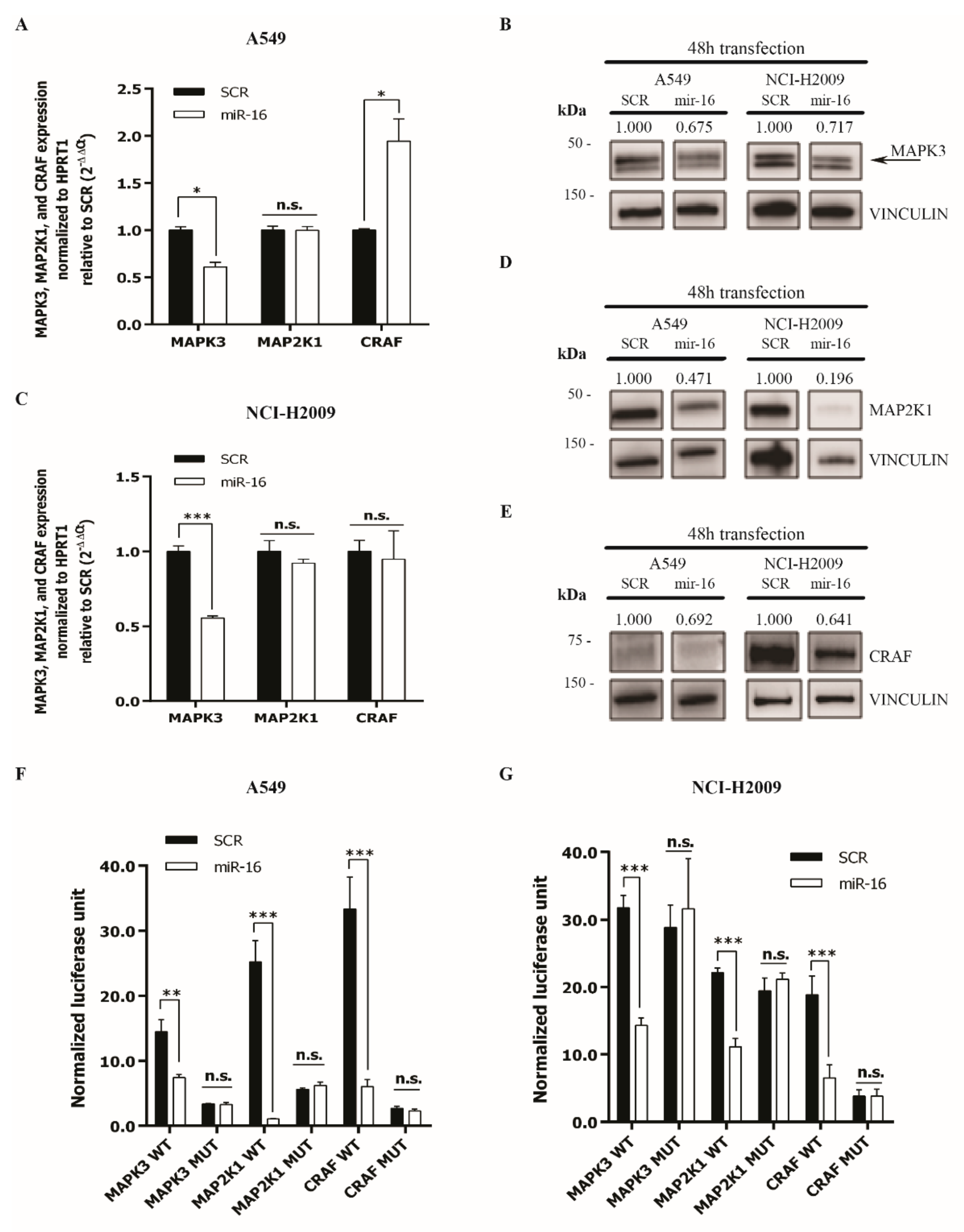
2.2. MicroRNA-16 Directly Targets MAPK3, MAP2K1, and CRAF
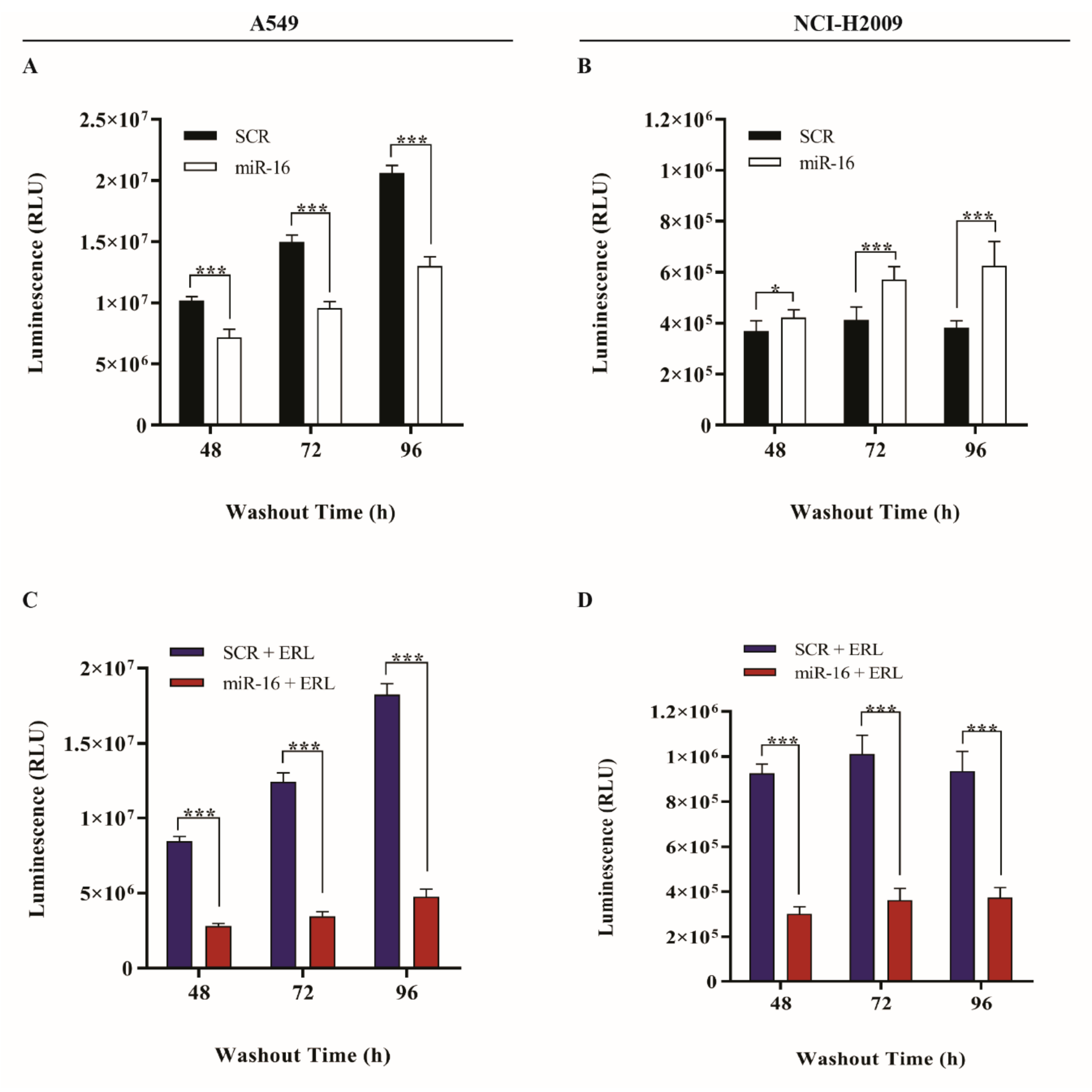
2.3. MicroRNA-16 Restores Sensitivity to Erlotinib in KRAS-Mutated NSCLC Cell Lines
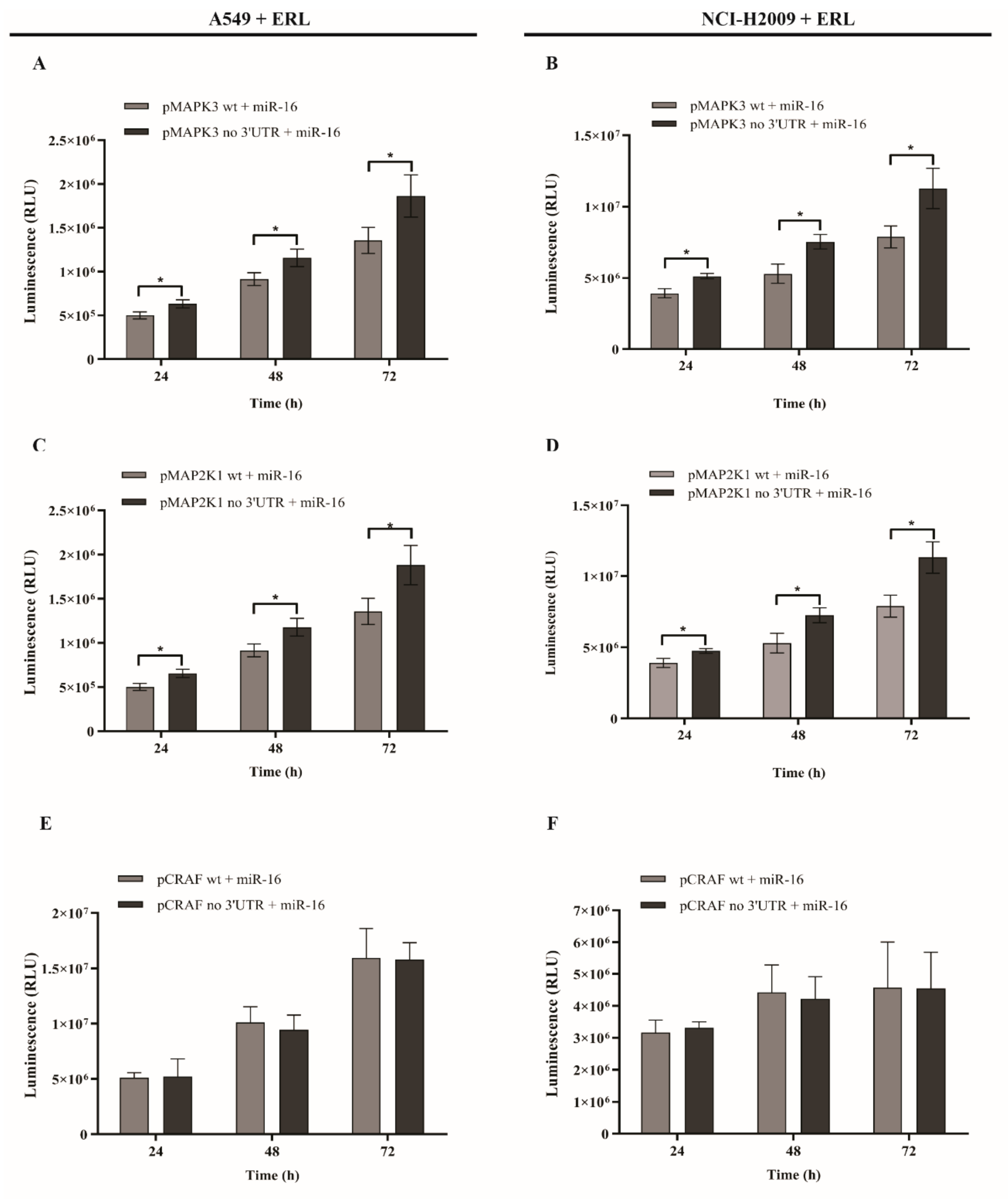
2.4. MicroRNA-16 Restores Sensitivity to Erlotinib by Targeting MAPK3 and MAP2K1
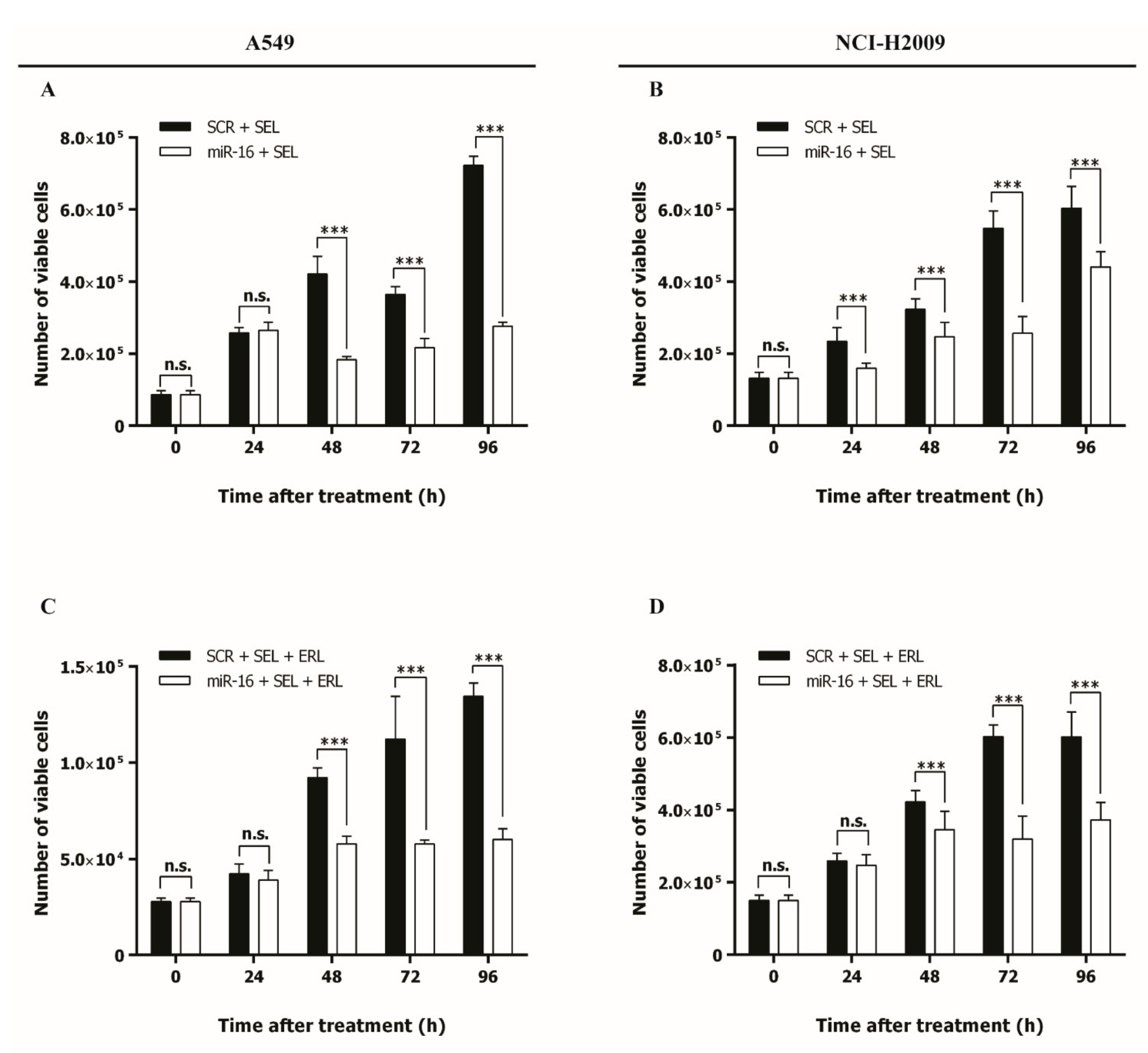
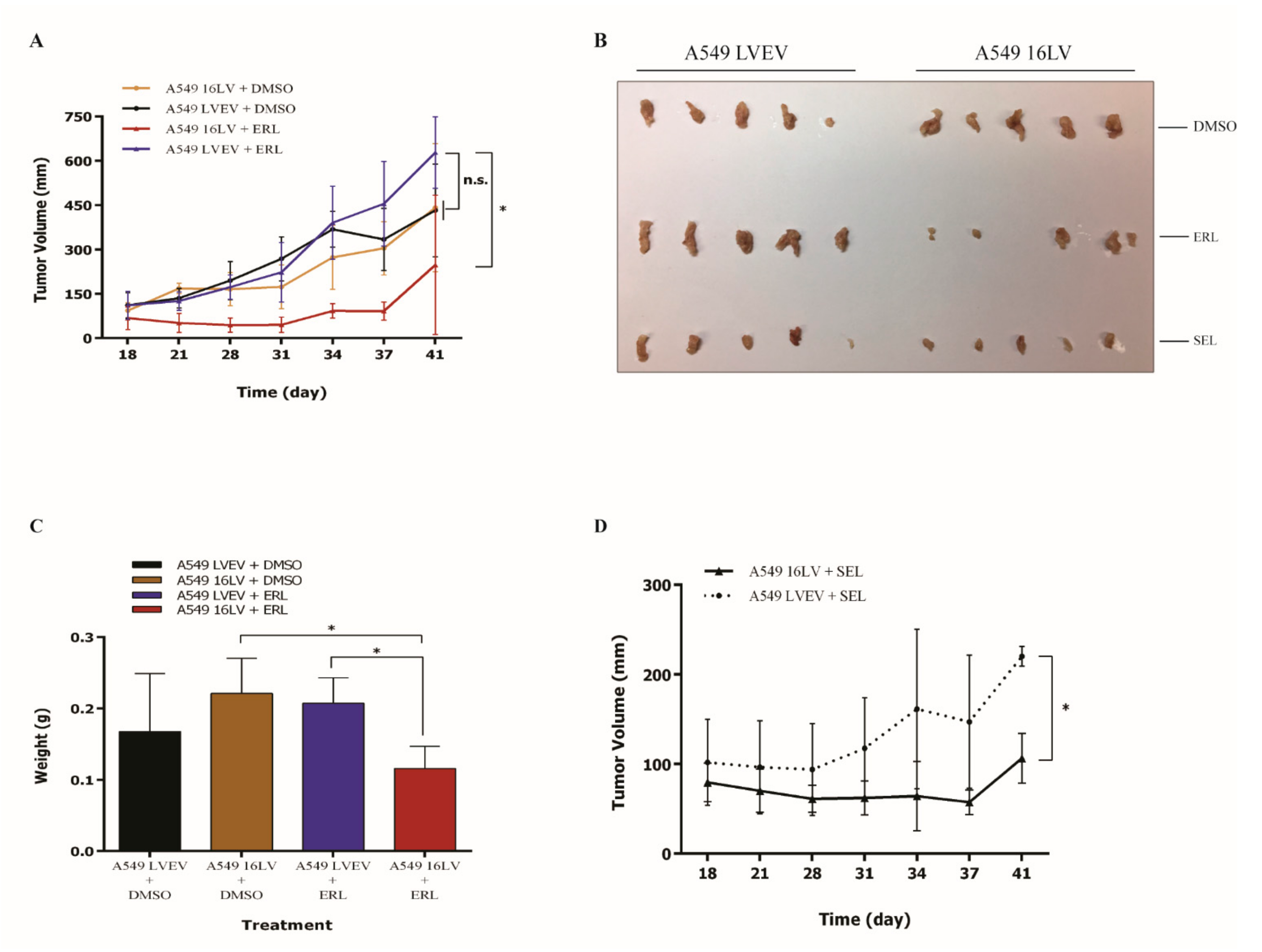
2.5. MicroRNA-16 Outperforms Selumetinib in Reducing NSCLC Growth In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell lines and Cell Cultures
4.3. Drugs
4.4. Transfection with microRNA Mimics
4.5. In Vitro Proliferation Assay
4.6. In Vitro Cell Growth Assays
- 4 h post-transfection with erlotinib at a single concentration of 1 μg/mL for 6 h and, following a medium change, allowed for 24–48–72–96 h washout;
- 10 h post-transfection with selumetinib at a single concentration of 1 μM continuously for 24–48–72–96 h.
4.7. In Vitro Cell Migration Assays
- 4 h post-transfection with erlotinib at a single concentration of 1 μg/mL for 6 h and, following a medium change, allowed for 24–48 h washout;
- 10 h post-transfection after erlotinib treatment with selumetinib at a single concentration of 1 μM continuously for 24–48 h.
4.8. DNA, RNA, and Protein Extraction
4.9. Analysis of KRAS Mutation Status
4.10. Evaluation of miRNAs and mRNA Expression
4.11. Protein Expression Analysis
4.12. Luciferase Assay
4.13. CellTiter-Glo® Luminescent Cell Viability Assay
4.14. Annexin-V Assay
4.15. Bromodeoxyuridine (BrdU) Assay
4.16. Animal Models
4.17. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | AKT serine/threonine kinase |
| CMS | Medical scientific committee |
| CRAF | Raf-1 proto-oncogene, serine/threonine kinase |
| DMSO | Dimethyl sulfoxide |
| EGFR | Epidermal growth factor receptor |
| ERL | Erlotinib |
| FBS | Fetal bovine serum |
| KRAS | KRAS proto-oncogene, GTPase |
| LV16 | LV16 |
| LVEV | Lentiviral empty vector |
| MAP2K1 | Mitogen-activated protein kinase kinase 1 |
| MAPK | Mitogen-activated protein kinases |
| MAPK3 | Mitogen-activated protein kinase 3 |
| MEK | Mitogen-activated protein kinase kinase |
| NSCLC | Non-small cell lung cancer |
| PI3K | Phosphoinositide 3-chinasi |
| PMSF | Phenylmethylsulphonyl fluoride |
| Pre-miRNA | miRNA precursor |
| SCR | Scrambled negative control |
| SEL | Selumetinib |
| TKIs | Tyrosine kinase inhibitors |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for Epidermal Growth Factor Receptor Mutations in Lung Cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Jänne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-Line Gefitinib in Patients With Advanced Non–Small-Cell Lung Cancer Harboring Somatic EGFR Mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef]
- Keedy, V.L.; Temin, S.; Somerfield, M.R.; Beasley, M.B.; Johnson, D.H.; McShane, L.M.; Milton, D.T.; Strawn, J.R.; Wakelee, H.A.; Giaccone, G. American Society of Clinical Oncology Provisional Clinical Opinion: Epidermal Growth Factor Receptor ( EGFR ) Mutation Testing for Patients With Advanced Non–Small-Cell Lung Cancer Considering First-Line EGFR Tyrosine Kinase Inhibitor Therapy. J. Clin. Oncol. 2011, 29, 2121–2127. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008, 9, 962–972. [Google Scholar] [CrossRef]
- Steckel, M.; Molina-Arcas, M.; Weigelt, B.; Marani, M.; Warne, P.H.; Kuznetsov, H.; Kelly, G.; Saunders, B.; Howell, M.; Downward, J.; et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012, 22, 1227–1245. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin. Cancer Res. 2015, 21, 1802–1809. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Carter, C.A.; Rajan, A.; Keen, C.; Szabo, E.; Khozin, S.; Thomas, A.; Brzezniak, C.; Guha, U.; Doyle, L.A.; Steinberg, S.M.; et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann. Oncol. 2016, 27, 693–699. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar]
- Iorio, M.V.; Croce, C.M. Causes and Consequences of MicroRNA Dysregulation. Cancer J. 2012, 18, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Vannini, I.; Fanini, F.; Fabbri, M. MicroRNAs as lung cancer biomarkers and key players in lung carcinogenesis. Clin. Biochem. 2013, 46, 918–925. [Google Scholar] [CrossRef]
- Valeri, N.; Vannini, I.; Fanini, F.; Calore, F.; Adair, B.; Fabbri, M. Epigenetics, miRNAs, and human cancer: A new chapter in human gene regulation. Mamm. Genome 2009, 20, 573–580. [Google Scholar] [CrossRef]
- Fabbri, M. miRNAs as molecular biomarkers of cancer. Expert Rev. Mol. Diagn. 2010, 10, 435–444. [Google Scholar] [CrossRef]
- Fanini, F.; Vannini, I.; Amadori, D.; Fabbri, M. Clinical Implications of MicroRNAs in Lung Cancer. Semin. Oncol. 2011, 38, 776–780. [Google Scholar] [CrossRef]
- Fabbri, M.; Calin, G.A. Beyond genomics: Interpreting the 93% of the human genome that does not encode proteins. Curr. Opin. Drug Discov. Devel. 2010, 13, 350–358. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Megraw, M.; Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. miRGen: A database for the study of animal microRNA genomic organization and function. Nucleic Acids Res. 2007, 35, D149–D155. [Google Scholar] [CrossRef] [Green Version]
- Alexiou, P.; Vergoulis, T.; Gleditzsch, M.; Prekas, G.; Dalamagas, T.; Megraw, M.; Grosse, I.; Sellis, T.; Hatzigeorgiou, A.G. miRGen 2.0: A database of microRNA genomic information and regulation. Nucleic Acids Res. 2010, 38, D137–D141. [Google Scholar] [CrossRef] [Green Version]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS Mutations and Primary Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, D.M.; Groffen, J. Four human carcinoma cell lines with novel mutations in position 12 of c-K-ras oncogene. Nucleic Acids Res. 1986, 14, 843–852. [Google Scholar]
- Yu, J.A.; Li, H.; Meng, X.; Fullerton, D.A.; Nemenoff, R.A.; Mitchell, J.D.; Weyant, M.J. Group IIa secretory phospholipase expression correlates with group IIa secretory phospholipase inhibition–mediated cell death in K-ras mutant lung cancer cells. J. Thorac. Cardiovasc. Surg. 2012, 144, 1479–1485. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.R.; Logie, A.; McKay, J.S.; Martin, P.; Steele, S.; Jenkins, R.; Cockerill, M.; Cartlidge, S.; Smith, P.D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: Mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical. Mol. Cancer Ther. 2007, 6, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Jazieh, A.-R.; Al Sudairy, R.; Abu-Shraie, N.; Al Suwairi, W.; Ferwana, M.; Murad, M.H. Erlotinib in wild type epidermal growth factor receptor non-small cell lung cancer: A systematic review. Ann. Thorac. Med. 2013, 8, 204–208. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Maemondo, M.; Ishii, Y.; Okudera, K.; Demura, Y.; Takamura, K.; Kobayashi, K.; Morikawa, N.; Gemma, A.; Ishimoto, O.; et al. A phase II study of erlotinib monotherapy in pre-treated non-small cell lung cancer without EGFR gene mutation who have never/light smoking history: Re-evaluation of EGFR gene status (NEJ006/TCOG0903). Lung Cancer 2014, 86, 195–200. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS Is Regulated by the let-7 MicroRNA Family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Bemis, L.T.; Nakajima, E.; Sugita, M.; Birks, D.K.; Robinson, W.A.; Varella-Garcia, M.; Bunn, P.A.; Haney, J.; Helfrich, B.A.; et al. EGFR regulation by microRNA in lung cancer: Correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann. Oncol. 2008, 19, 1053–1059. [Google Scholar] [CrossRef]
- Webster, R.J.; Giles, K.M.; Price, K.J.; Zhang, P.M.; Mattick, J.S.; Leedman, P.J. Regulation of Epidermal Growth Factor Receptor Signaling in Human Cancer Cells by MicroRNA-7. J. Biol. Chem. 2009, 284, 5731–5741. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Tao, T.; Wang, Y.; Fang, F.; Huang, Y.; Chen, S.; Zhu, W.; Chen, M. hsa-miR-135a-1 inhibits prostate cancer cell growth and migration by targeting EGFR. Tumor Biol. 2016, 37, 14141–14151. [Google Scholar] [CrossRef]
- Wang, F.; Meng, F.; Wang, L.; Wong, S.C.C.; Cho, W.C.S.; Chan, L.W.C. Associations of mRNA:microRNA for the Shared Downstream Molecules of EGFR and Alternative Tyrosine Kinase Receptors in Non-small Cell Lung Cancer. Front. Genet. 2016, 7, 173. [Google Scholar] [CrossRef]
- Meng, F.; Wang, F.; Wang, L.; Wong, S.C.C.; Cho, W.C.S.; Chan, L.W.C. MiR-30a-5p Overexpression May Overcome EGFR-Inhibitor Resistance through Regulating PI3K/AKT Signaling Pathway in Non-small Cell Lung Cancer Cell Lines. Front. Genet. 2016, 7, 197. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, J.; Sun, Y.; Zhang, Y.; Dong, L.; Shen, C.; Yang, L.; Yang, M.; Li, Y.; Shen, G.; et al. miR-7 reverses the resistance to BRAFi in melanoma by targeting EGFR/IGF-1R/CRAF and inhibiting the MAPK and PI3K/AKT signaling pathways. Oncotarget 2016, 7, 53558. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. Downregulation of miR-16 promotes growth and motility by targeting HDGF in non-small cell lung cancer cells. FEBS Lett. 2013, 587, 3153–3157. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zhang, D.; Du, R.; Pan, Y.; Zhao, L.; Sun, S.; Hong, L.; Liu, J.; Fan, D. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int. J. Cancer 2008, 123, 372–379. [Google Scholar] [CrossRef]
- Cittelly, D.M.; Das, P.M.; Salvo, V.A.; Fonseca, J.P.; Burow, M.E.; Jones, F.E. Oncogenic HER2Δ16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis 2010, 31, 2049–2057. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Chattopadhyay, D.; Chakrabarti, G. miR-16 targets Bcl-2 in paclitaxel-resistant lung cancer cells and overexpression of miR-16 along with miR-17 causes unprecedented sensitivity by simultaneously modulating autophagy and apoptosis. Cell. Signal. 2015, 27, 189–203. [Google Scholar] [CrossRef]
- Han, J.; Chen, Q. MiR-16 modulate temozolomide resistance by regulating BCL-2 in human glioma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 12698–12707. [Google Scholar]
- You, C.; Liang, H.; Sun, W.; Li, J.; Liu, Y.; Fan, Q.; Zhang, H.; Yue, X.; Li, J.; Chen, X.; et al. Deregulation of the miR-16-KRAS axis promotes colorectal cancer. Sci. Rep. 2016, 6, 37459. [Google Scholar] [CrossRef] [Green Version]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro Cell Migration and Invasion Assays. J. Vis. Exp. 2014, 88, 51046. [Google Scholar] [CrossRef] [Green Version]
- Krypuy, M.; Newnham, G.M.; Thomas, D.M.; Conron, M.; Dobrovic, A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer 2006, 6, 295. [Google Scholar]
- Notta, F.; Doulatov, S.; Dick, J.E. Engraftment of human hematopoietic stem cells is more efficient in female NOD/SCID/IL-2Rgc-null recipients. Blood 2010, 115, 3704–3707. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanini, F.; Bandini, E.; Plousiou, M.; Carloni, S.; Wise, P.; Neviani, P.; Murtadha, M.; Foca, F.; Fabbri, F.; Vannini, I.; et al. MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 13357. https://doi.org/10.3390/ijms222413357
Fanini F, Bandini E, Plousiou M, Carloni S, Wise P, Neviani P, Murtadha M, Foca F, Fabbri F, Vannini I, et al. MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2021; 22(24):13357. https://doi.org/10.3390/ijms222413357
Chicago/Turabian StyleFanini, Francesca, Erika Bandini, Meropi Plousiou, Silvia Carloni, Petra Wise, Paolo Neviani, Mariam Murtadha, Flavia Foca, Francesco Fabbri, Ivan Vannini, and et al. 2021. "MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 22, no. 24: 13357. https://doi.org/10.3390/ijms222413357
APA StyleFanini, F., Bandini, E., Plousiou, M., Carloni, S., Wise, P., Neviani, P., Murtadha, M., Foca, F., Fabbri, F., Vannini, I., & Fabbri, M. (2021). MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 22(24), 13357. https://doi.org/10.3390/ijms222413357