
The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A

 , , ,
, , ,  and add
Show full author list
and add
Show full author list
Abstract
:1. Introduction
2. Results
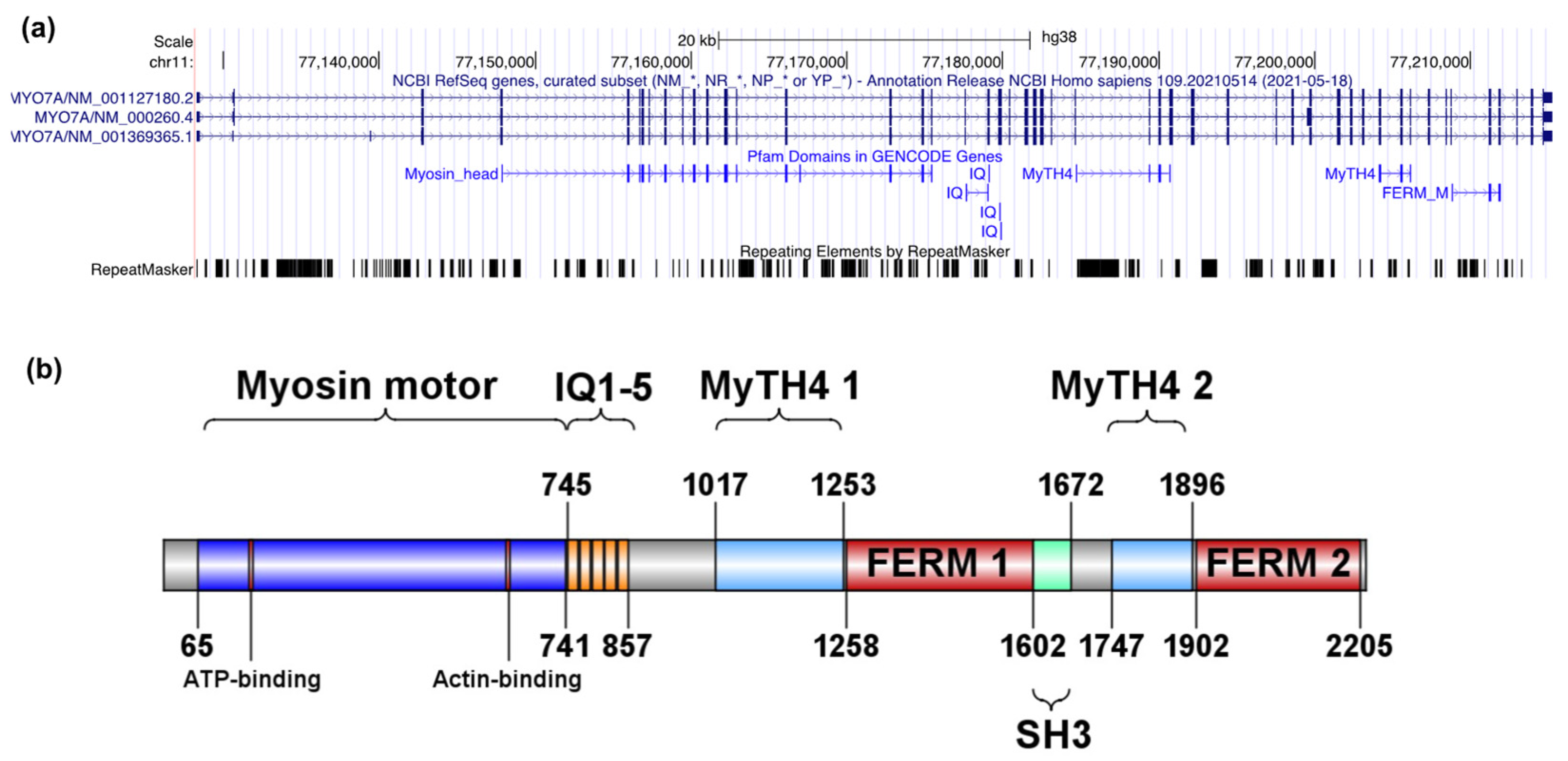
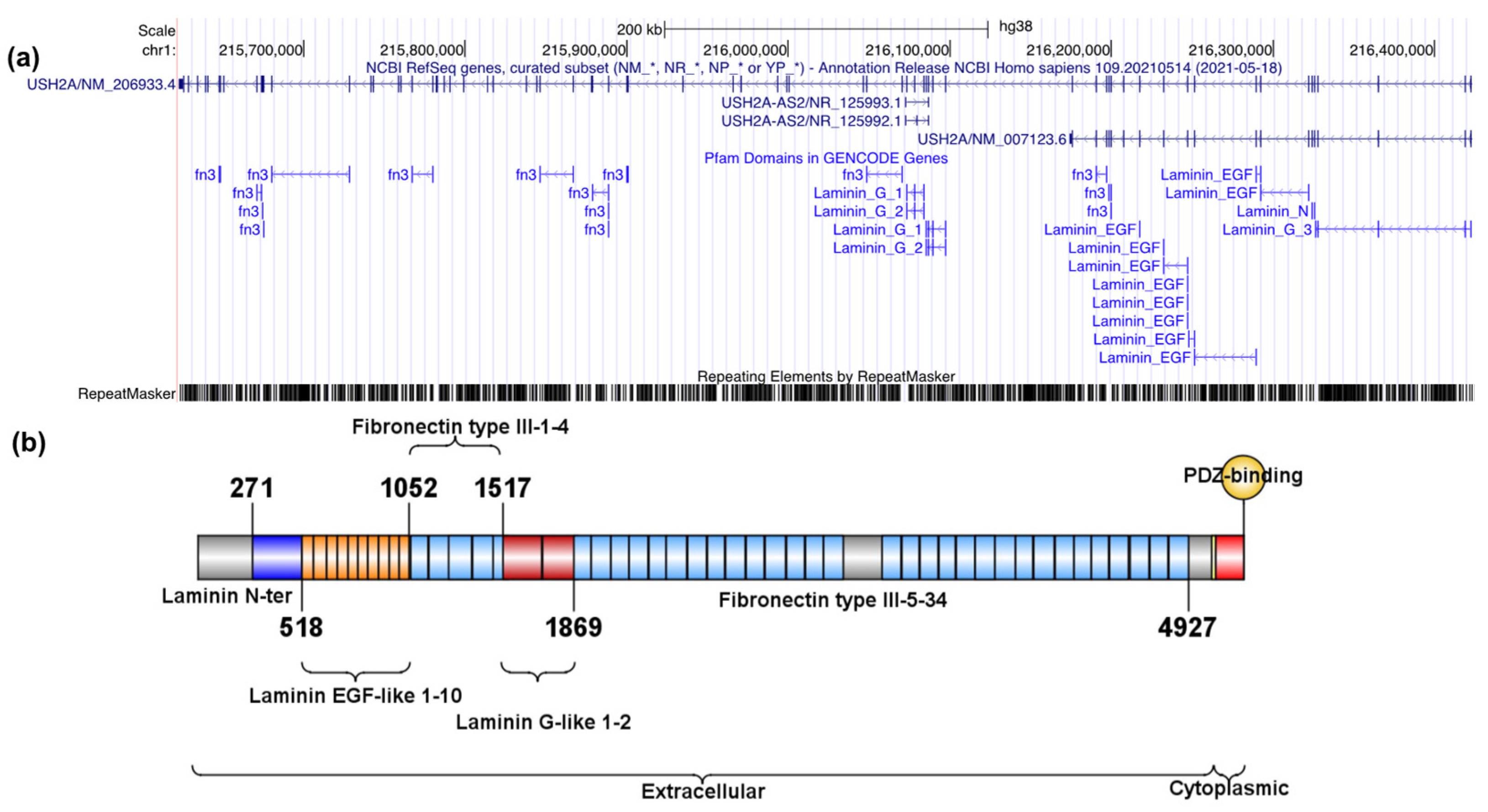
2.1. MYO7A and USH2A Variant Identification and Spectrum
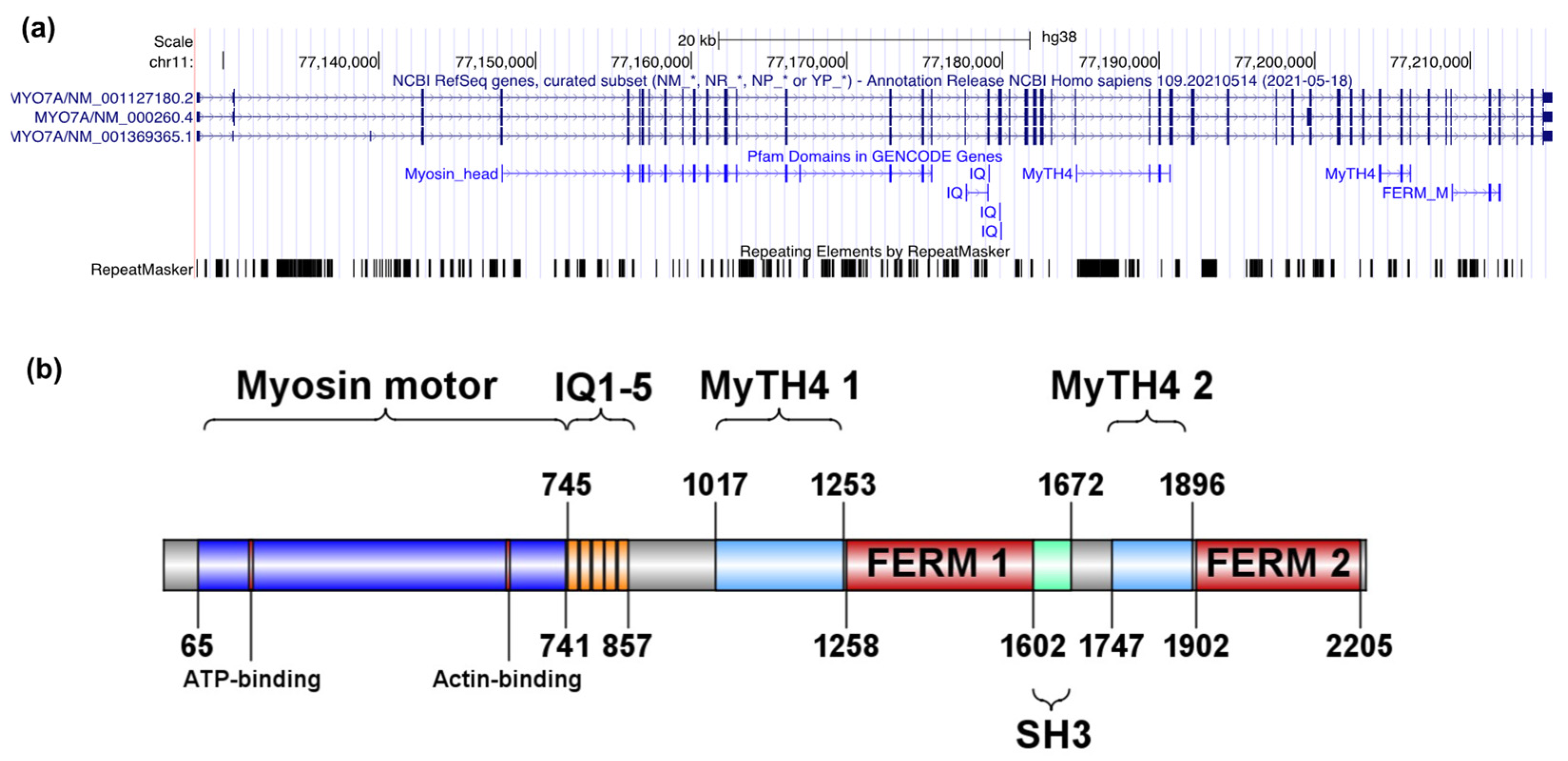
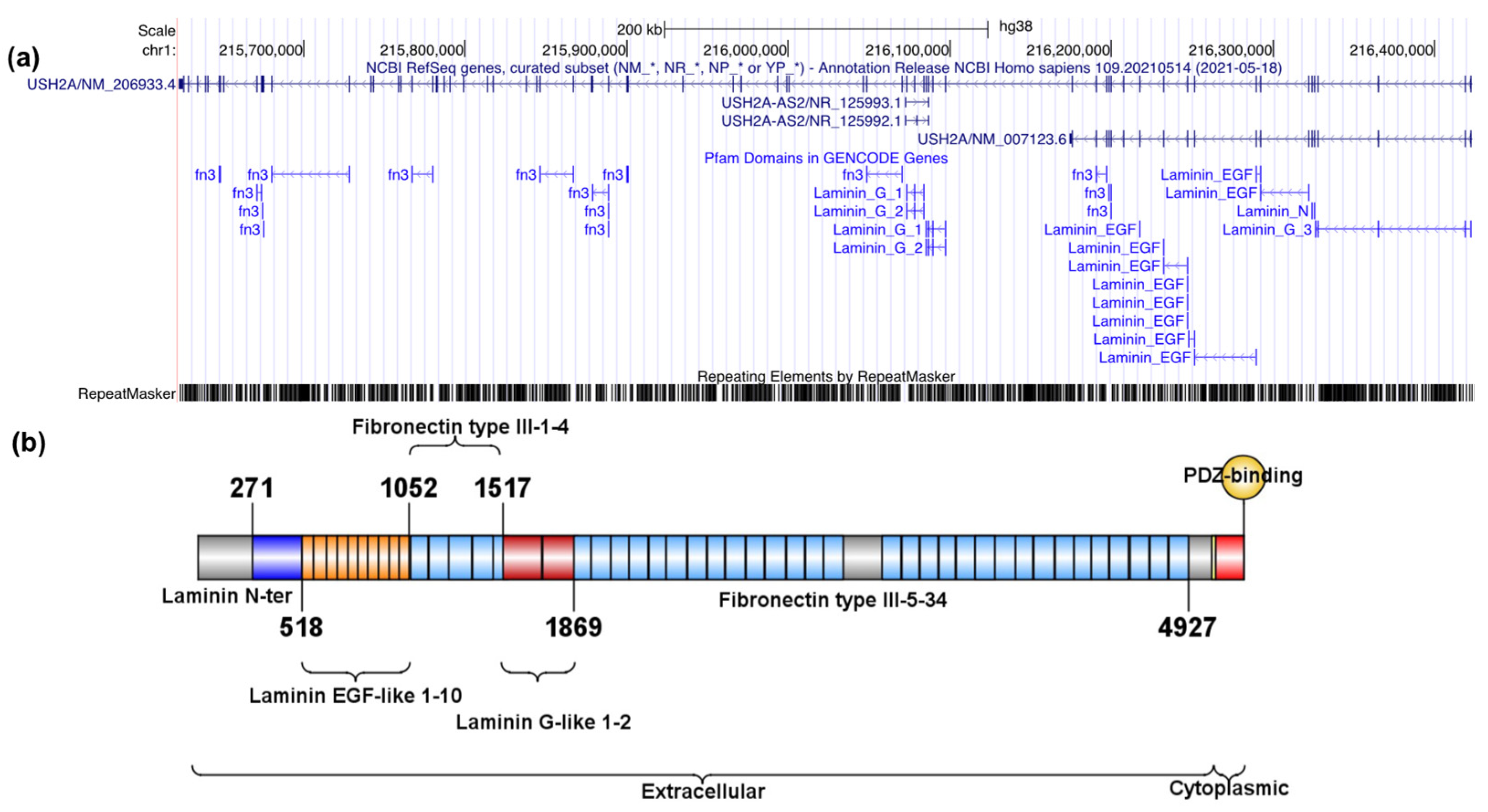
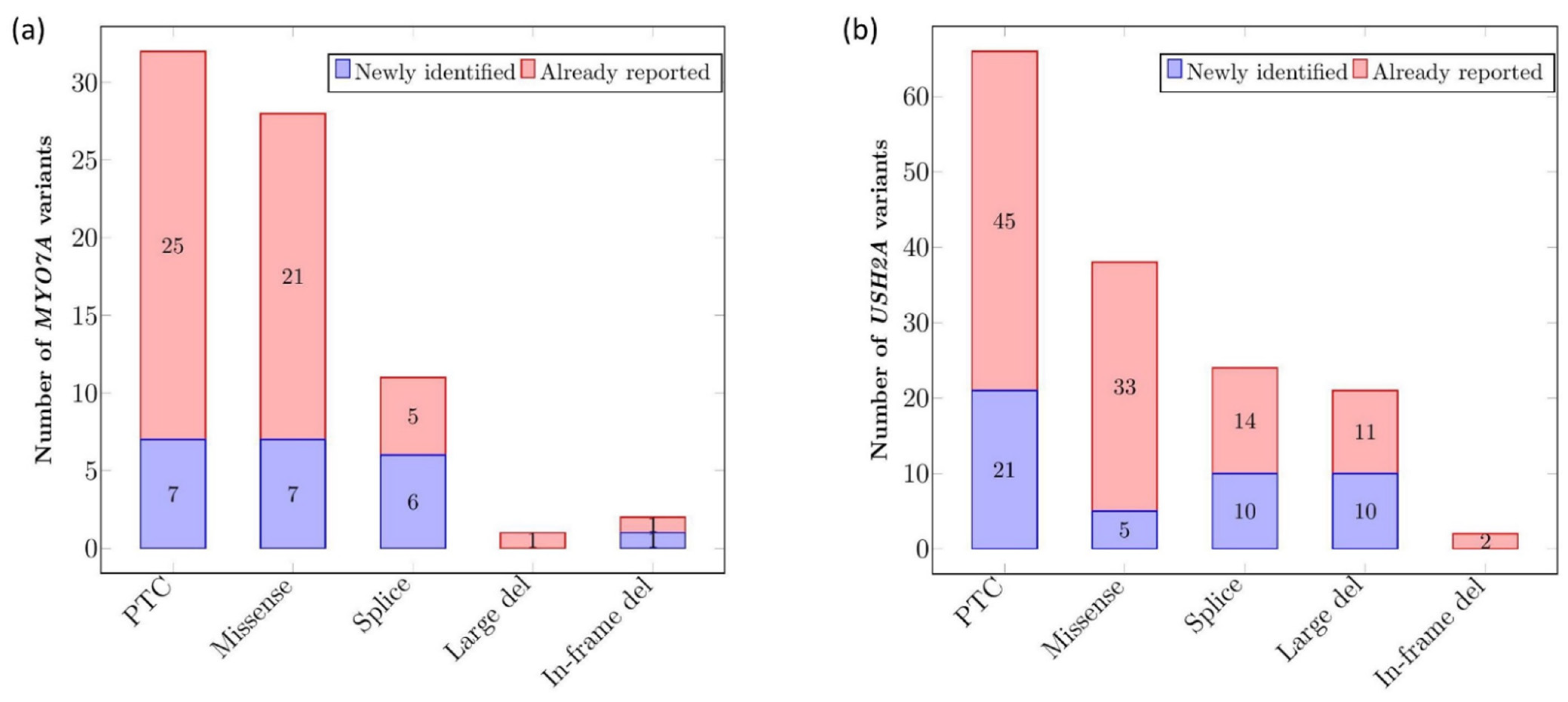
2.1.1. Description of MYO7A and USH2A Alterations
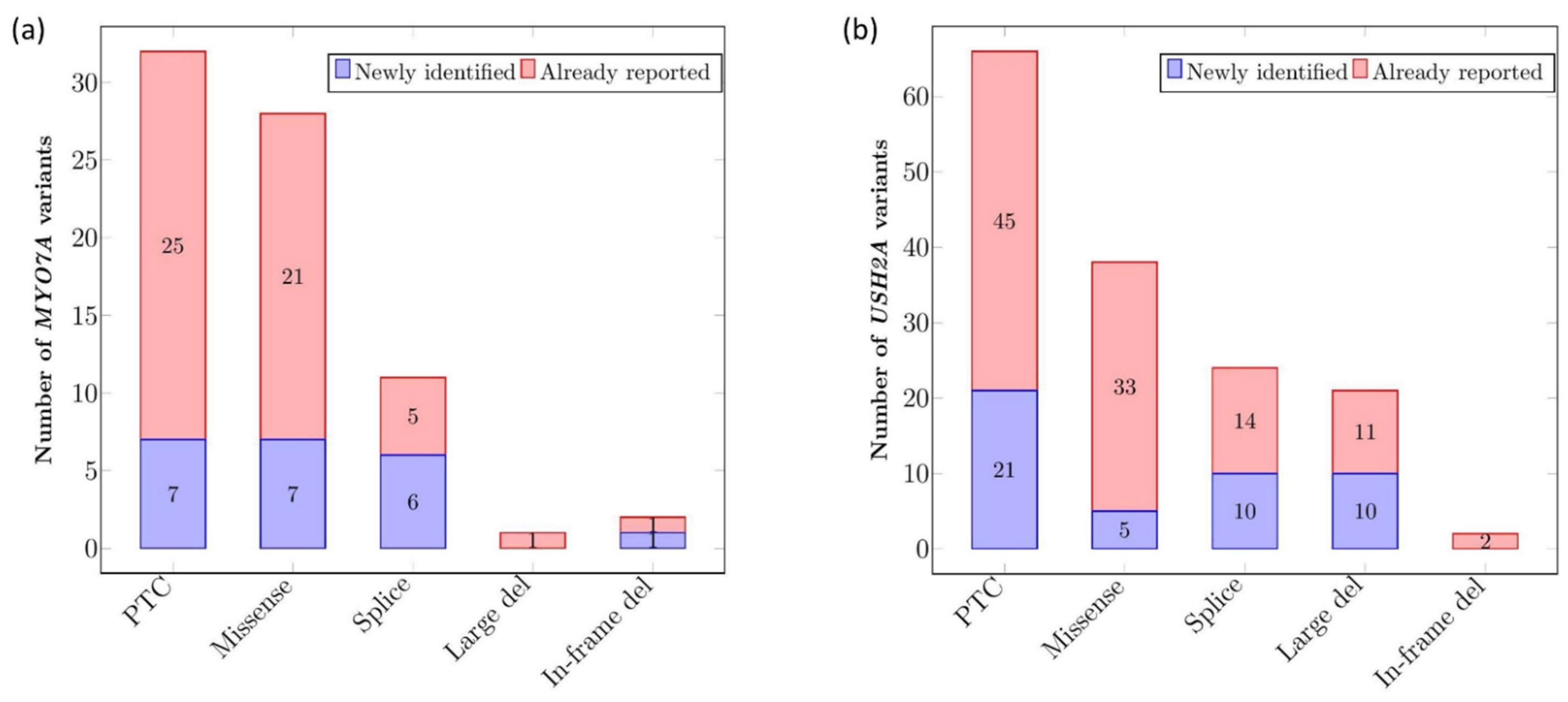
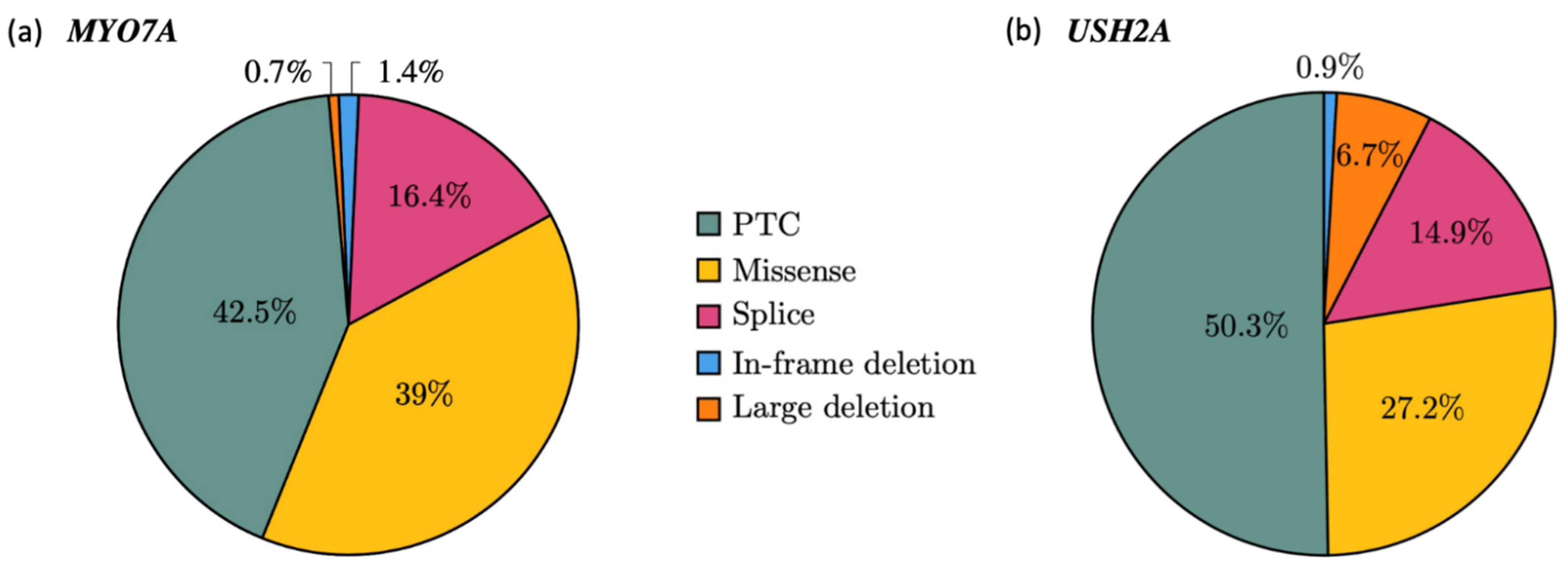
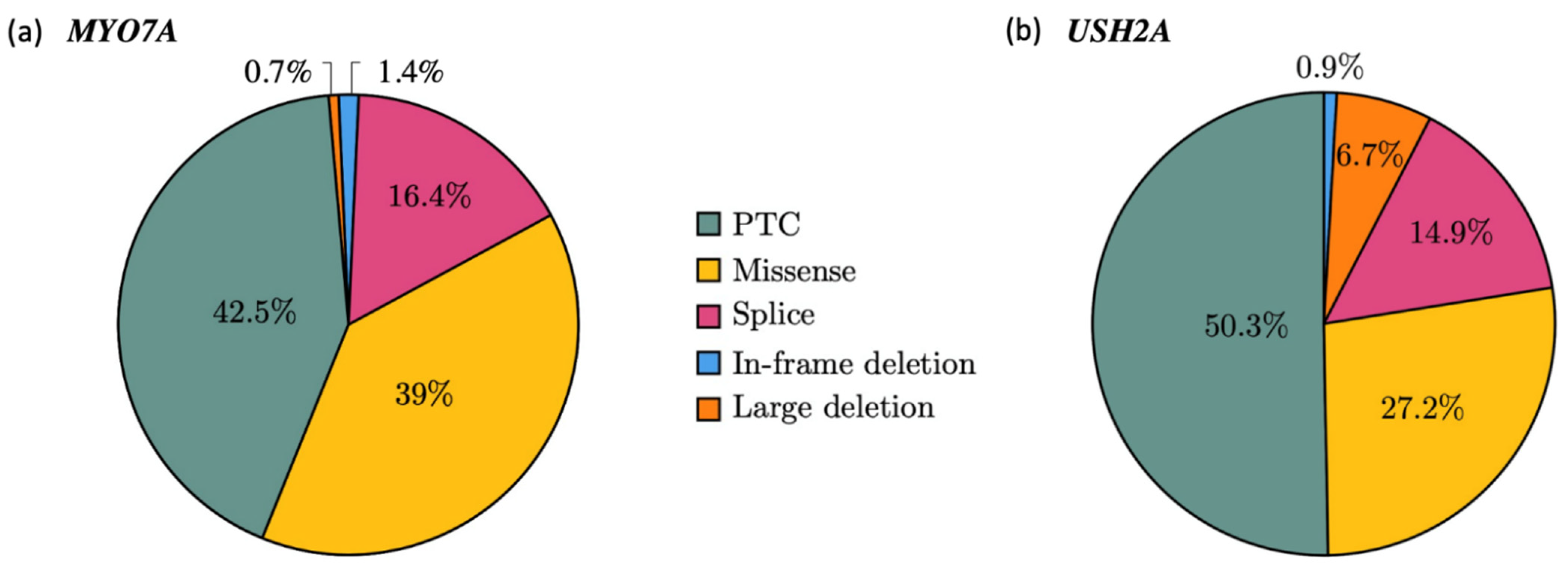
2.1.2. Mutational Spectrum of the Cohort
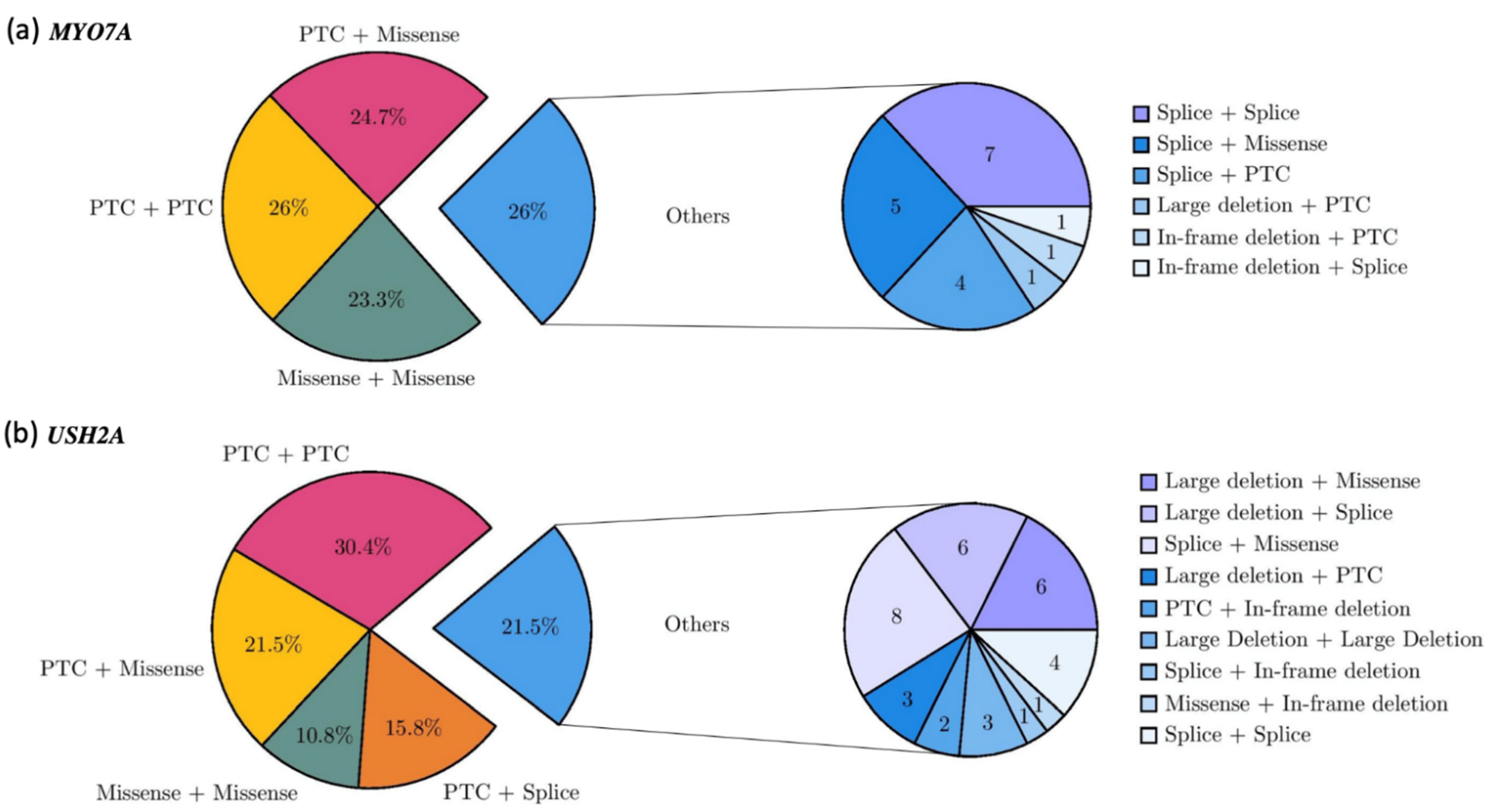
2.1.3. Recurrence of Alterations
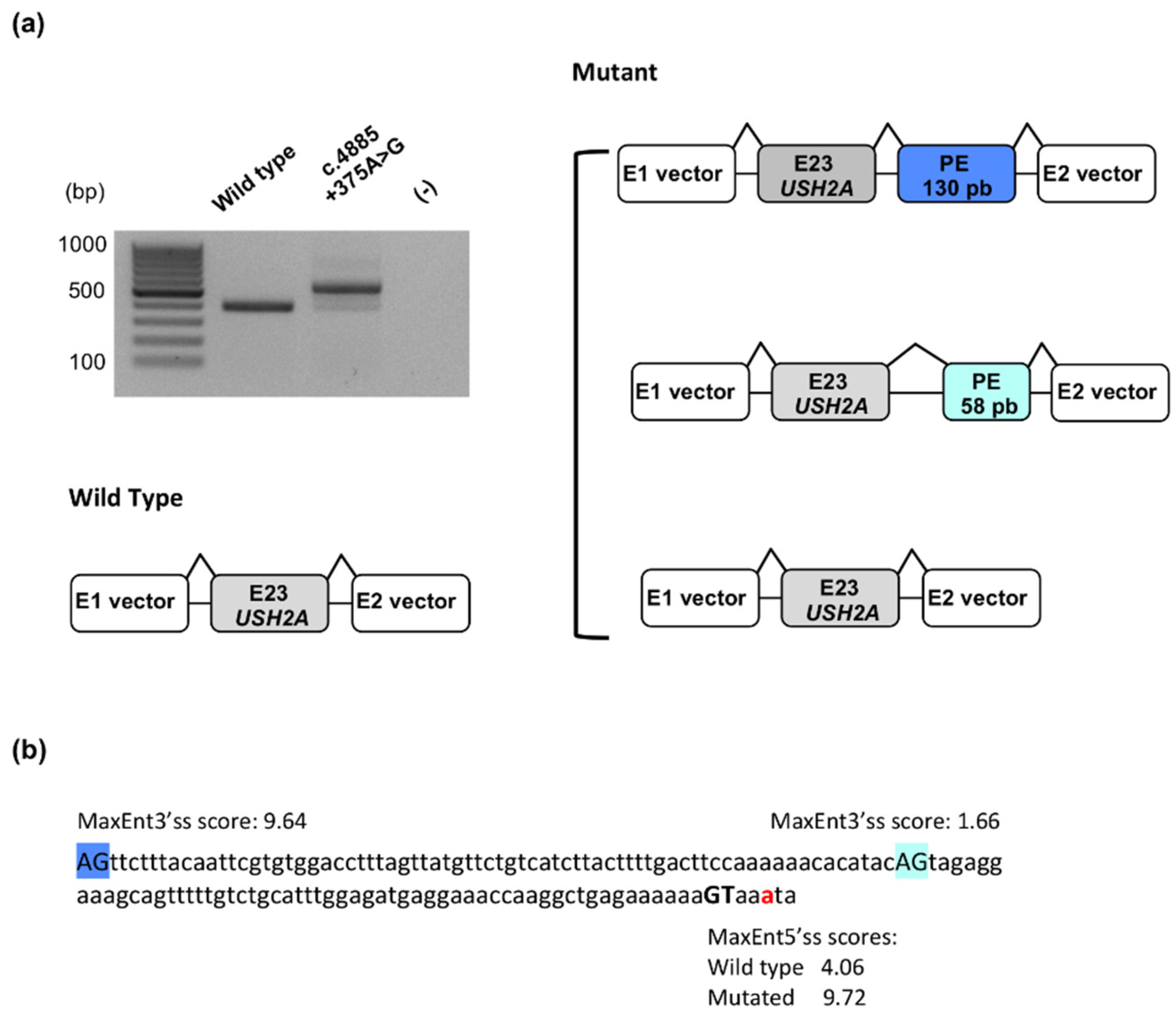
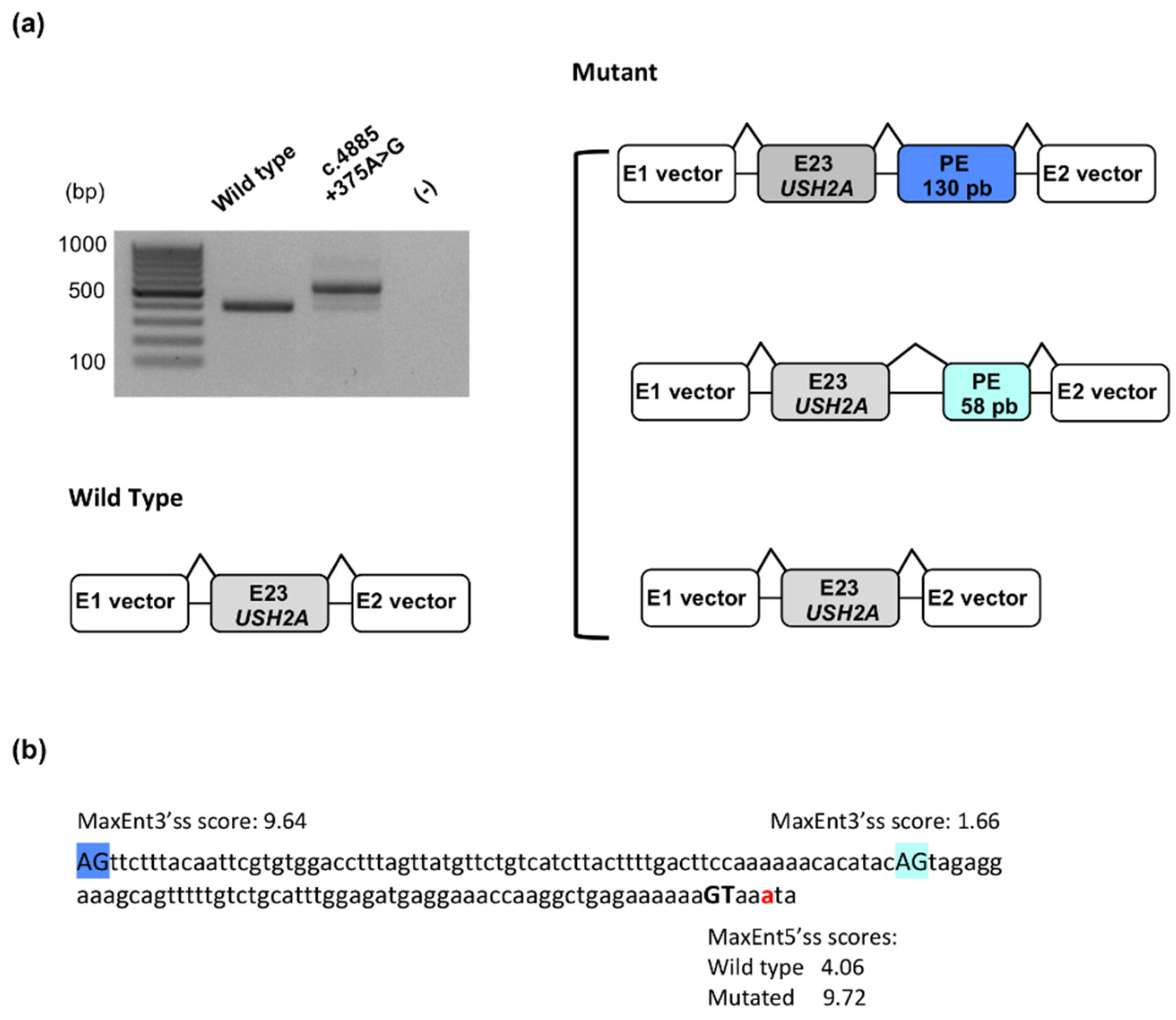
2.1.4. Identification of a Novel Deep Intronic Variation
2.2. Spectrum of MYO7A and USH2A Pathogenic Genotypes
2.2.1. Pathogenic Classification of MYO7A and USH2A Genotypes
2.2.2. Genotype Spectrum
2.2.3. Additional Pathogenic Variant in a Third USH Gene
2.2.4. Clinical Evaluation
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment
4.2. Molecular Analyses
4.2.1. Gene-Panel Sequencing and Bioinformatics
4.2.2. Complementary Analysis
4.3. Pathogenicity Assessment
4.4. Functional Analysis
4.5. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evidence of Pathogenicity | Description |
|---|---|
| Very strong | PVS1 null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function is a known mechanism of disease |
| Strong | PS1 Same amino acid change as a previously established pathogenic variant regardless of nucleotide change |
| PS2 De novo (both maternity and paternity confirmed) in a patient with the disease and no family history | |
| PS3 Well-establishedin vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product | |
| PS4 The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls | |
| Moderate | PM1 Located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation |
| PM2 Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or the Genome Aggregation Database | |
| PM3 For recessive disorders, detected in trans with a pathogenic variant | |
| PM4 Protein length changes as a result of in-frame deletions/insertions in a nonrepeat region or stop-loss variants | |
| PM5 Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before | |
| PM6 Assumed de novo, but without confirmation of paternity and maternity | |
| Supporting | PP1 Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease |
| PP2 Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease | |
| PP3 Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) | |
| PP4 Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | |
| PP5 Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation |
References
- Millán, J.M.; Aller, E.; Jaijo, T.; Blanco-Kelly, F.; Gimenez-Pardo, A.; Ayuso, C. An Update on the Genetics of Usher Syndrome. J. Ophthalmol. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Le Quesne Stabej, P.; Saihan, Z.; Rangesh, N.; Steele-Stallard, H.B.; Ambrose, J.; Coffey, A.; Emmerson, J.; Haralambous, E.; Hughes, Y.; Steel, K.P.; et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012, 49, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Roux, A.-F.; Faugère, V.; Vaché, C.; Baux, D.; Besnard, T.; Léonard, S.; Blanchet, C.; Hamel, C.; Mondain, M.; Gilbert-Dussardier, B.; et al. Four-Year Follow-up of Diagnostic Service in USH1 Patients. Investig. Opthalmol. Vis. Sci. 2011, 52, 4063–4071. [Google Scholar] [CrossRef]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.-P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Abadie, C.; Blanchet, C.; Baux, D.; Larrieu, L.; Besnard, T.; Ravel, P.; Biboulet, R.; Hamel, C.; Malcolm, S.; Mondain, M.; et al. Audiological findings in 100 USH2 patients. Clin. Genet. 2011, 82, 433–438. [Google Scholar] [CrossRef]
- Besnard, T.; Vaché, C.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Odent, S.; Blanchet, P.; Calvas, P.; Hamel, C.; et al. Non-USH2A mutations in USH2 patients. Hum. Mutat. 2012, 33, 504–510. [Google Scholar] [CrossRef]
- García-García, G.; Besnard, T.; Baux, D.; Vaché, C.; Aller, E.; Malcolm, S.; Claustres, M.; Millan, J.M.; Roux, A.-F. The contribution of GPR98 and DFNB31 genes to a Spanish Usher syndrome type 2 cohort. Mol. Vis. 2013, 19, 367–373. [Google Scholar]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kabahuma, R.; Schubert, W.-D.; Labuschagne, C.; Yan, D.; Blanton, S.; Pepper, M.; Liu, X. Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations. Genes 2021, 12, 274. [Google Scholar] [CrossRef]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands With Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Opthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-García, G.; Jaijo, T.; Fornés, N.; Ayuso, C.; Fernández-Burriel, M.; La Morena, A.S.-D.; Aller, E.; Millán, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef]
- Neuhaus, C.; Eisenberger, T.; Decker, C.; Nagl, S.; Blank, C.; Pfister, M.; Kennerknecht, I.; Müller-Hofstede, C.; Issa, P.C.; Heller, R.; et al. Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: Copy number variations, phenocopies, a predominant target for translational read-through, andPEX26mutated in Heimler syndrome. Mol. Genet. Genom. Med. 2017, 5, 531–552. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Vaché, C.; Besnard, T.; le Berre, P.; García-García, G.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Bolz, H.J.; Millan, J.; et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: Implications for diagnosis and therapy. Hum. Mutat. 2011, 33, 104–108. [Google Scholar] [CrossRef]
- Patel, M.J.; DiStefano, M.T.; Oza, A.M.; Hughes, M.Y.; Wilcox, E.H.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; et al. Disease-specific ACMG/AMP guidelines improve sequence variant interpretation for hearing loss. Genet. Med. 2021, 23, 2208–2212. [Google Scholar] [CrossRef]
- van der Sluijs, P.J.; Alders, M.; Dingemans, A.J.M.; Parbhoo, K.; van Bon, B.W.; Dempsey, J.C.; Doherty, D.; Dunnen, J.T.D.; Gerkes, E.H.; Milller, I.M.; et al. A Case Series of Familial ARID1B Variants Illustrating Variable Expression and Suggestions to Update the ACMG Criteria. Genes 2021, 12, 1275. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum. Mutat. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Davieson, C.D.; Joyce, K.E.; Sharma, L.; Shovlin, C.L. DNA variant classification–reconsidering “allele rarity” and “phenotype” criteria in ACMG/AMP guidelines. Eur. J. Med Genet. 2021, 64, 104312. [Google Scholar] [CrossRef]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2019, 22, 336–344. [Google Scholar] [CrossRef]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilf-Yarkoni, A.; Shor, O.; Fellner, A.; Hellmann, M.A.; Pras, E.; Yonath, H.; Shkedi-Rafid, S.; Basel-Salmon, L.; Bazak, L.; Eliahou, R.; et al. Mild Phenotype of Wolfram Syndrome Associated With a Common Pathogenic Variant Is Predicted by a Structural Model of Wolframin. Neurol. Genet. 2021, 7, e578. [Google Scholar] [CrossRef]
- Reurink, J.; Dockery, A.; Oziębło, D.; Farrar, G.; Ołdak, M.; Brink, J.T.; Bergen, A.; Rinne, T.; Yntema, H.; Pennings, R.; et al. Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases. Int. J. Mol. Sci. 2021, 22, 6419. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carro, R.; Blanco-Kelly, F.; Galbis-Martínez, L.; García-García, G.; Aller, E.; García-Sandoval, B.; Mínguez, P.; Corton, M.; Mahíllo-Fernández, I.; Martín-Mérida, I.; et al. Unravelling the pathogenic role and genotype-phenotype correlation of the USH2A p.(Cys759Phe) variant among Spanish families. PLoS ONE 2018, 13, e0199048. [Google Scholar] [CrossRef]
- Trapani, I.; Auricchio, A. Has retinal gene therapy come of age? From bench to bedside and back to bench. Hum. Mol. Genet. 2019, 28, R108–R118. [Google Scholar] [CrossRef] [Green Version]
- Zallocchi, M.; Binley, K.; Lad, Y.; Ellis, S.; Widdowson, P.; Iqball, S.; Scripps, V.; Kelleher, M.; Loader, J.; Miskin, J.; et al. EIAV-Based Retinal Gene Therapy in the shaker1 Mouse Model for Usher Syndrome Type 1B: Development of UshStat. PLoS ONE 2014, 9, e94272. [Google Scholar] [CrossRef] [Green Version]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef] [PubMed]
- Gerard, X.; Garanto, A.; Rozet, J.-M.; Collin, R.W.J. Antisense Oligonucleotide Therapy for Inherited Retinal Dystrophies. Adv. Exp. Med. Biol. 2016, 854, 517–524. [Google Scholar] [CrossRef]
- Liquori, A.; Vaché, C.; Baux, D.; Blanchet, C.; Hamel, C.; Malcolm, S.; Koenig, M.; Claustres, M.; Roux, A.-F. WholeUSH2AGene Sequencing Identifies Several New Deep Intronic Mutations. Hum. Mutat. 2015, 37, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Le Guédard-Méreuze, S.; Vaché, C.; Baux, D.; Faugère, V.; Larrieu, L.; Abadie, C.; Janecke, A.; Claustres, M.; Roux, A.-F.; Tuffery-Giraud, S. Ex vivo splicing assays of mutations at noncanonical positions of splice sites in USHER genes. Hum. Mutat. 2010, 31, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bottillo, I.; De Luca, A.; Schirinzi, A.; Guida, V.; Torrente, I.; Calvieri, S.; Gervasini, C.; Larizza, L.; Pizzuti, A.; Dallapiccola, B. Functional analysis of splicing mutations in exon 7 of NF1gene. BMC Med. Genet. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Baux, D.; Van Goethem, C.; Ardouin, O.; Guignard, T.; Bergougnoux, A.; Koenig, M.; Roux, A.-F. MobiDetails: Online DNA variants interpretation. Eur. J. Hum. Genet. 2020, 29, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Wenzhong, L.; Yubon, X.; Jiyong, M.; Xiaotong, L.; Peng, N.; Zhixiang, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansard, L.; Baux, D.; Vaché, C.; Blanchet, C.; Meunier, I.; Willems, M.; Faugère, V.; Baudoin, C.; Moclyn, M.; Bianchi, J.; et al. The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A. Int. J. Mol. Sci. 2021, 22, 13294. https://doi.org/10.3390/ijms222413294
Mansard L, Baux D, Vaché C, Blanchet C, Meunier I, Willems M, Faugère V, Baudoin C, Moclyn M, Bianchi J, et al. The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A. International Journal of Molecular Sciences. 2021; 22(24):13294. https://doi.org/10.3390/ijms222413294
Chicago/Turabian StyleMansard, Luke, David Baux, Christel Vaché, Catherine Blanchet, Isabelle Meunier, Marjolaine Willems, Valérie Faugère, Corinne Baudoin, Melody Moclyn, Julie Bianchi, and et al. 2021. "The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A" International Journal of Molecular Sciences 22, no. 24: 13294. https://doi.org/10.3390/ijms222413294
APA StyleMansard, L., Baux, D., Vaché, C., Blanchet, C., Meunier, I., Willems, M., Faugère, V., Baudoin, C., Moclyn, M., Bianchi, J., Dollfus, H., Gilbert-Dussardier, B., Dupin-Deguine, D., Bonneau, D., Drumare, I., Odent, S., Zanlonghi, X., Claustres, M., Koenig, M., ... Roux, A.-F. (2021). The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A. International Journal of Molecular Sciences, 22(24), 13294. https://doi.org/10.3390/ijms222413294