
A Novel Renoprotective Strategy: Upregulation of PD-L1 Mitigates Cisplatin-Induced Acute Kidney Injury

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
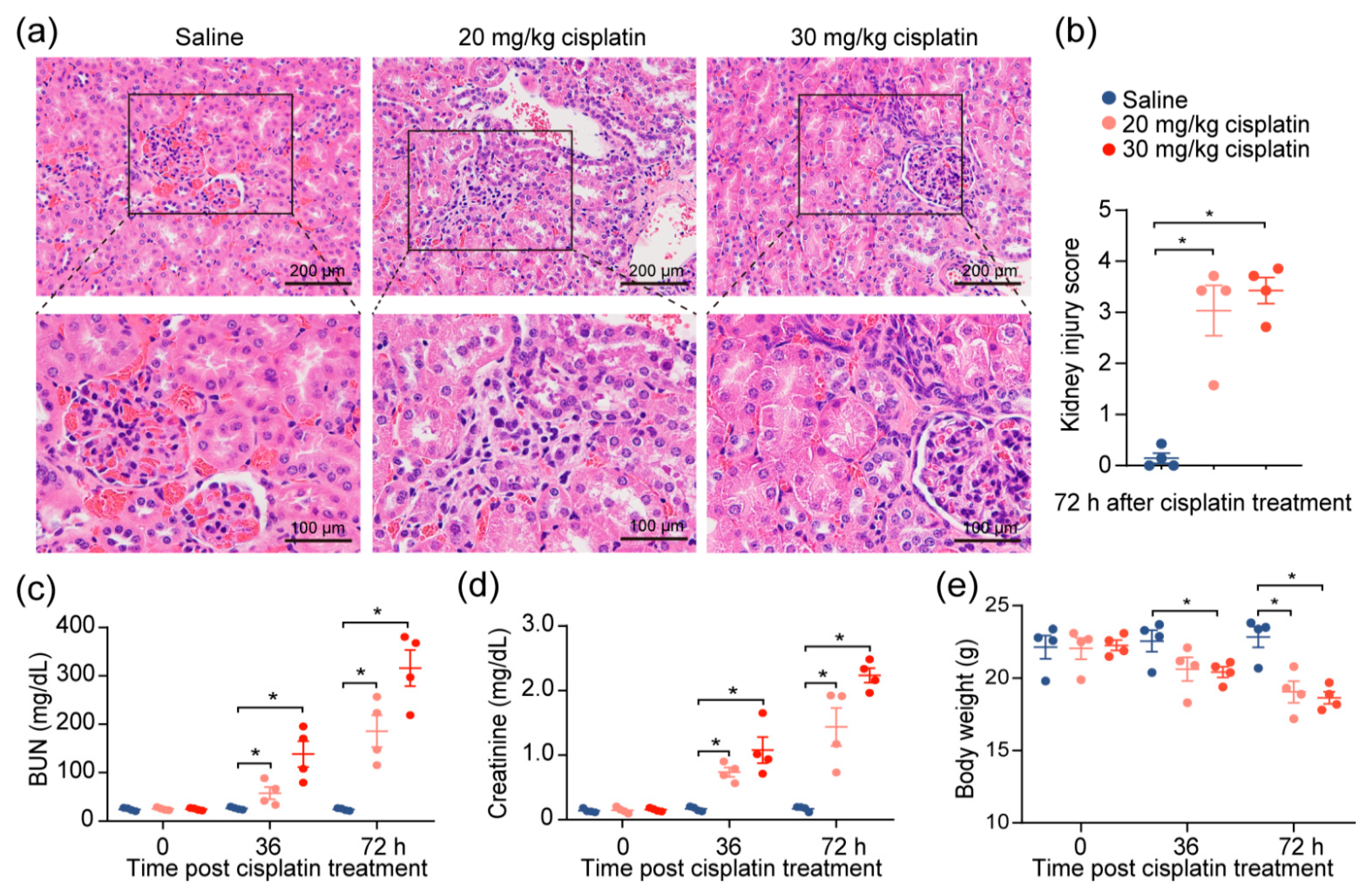
2.1. Cisplatin Induces Acute Kidney Injury
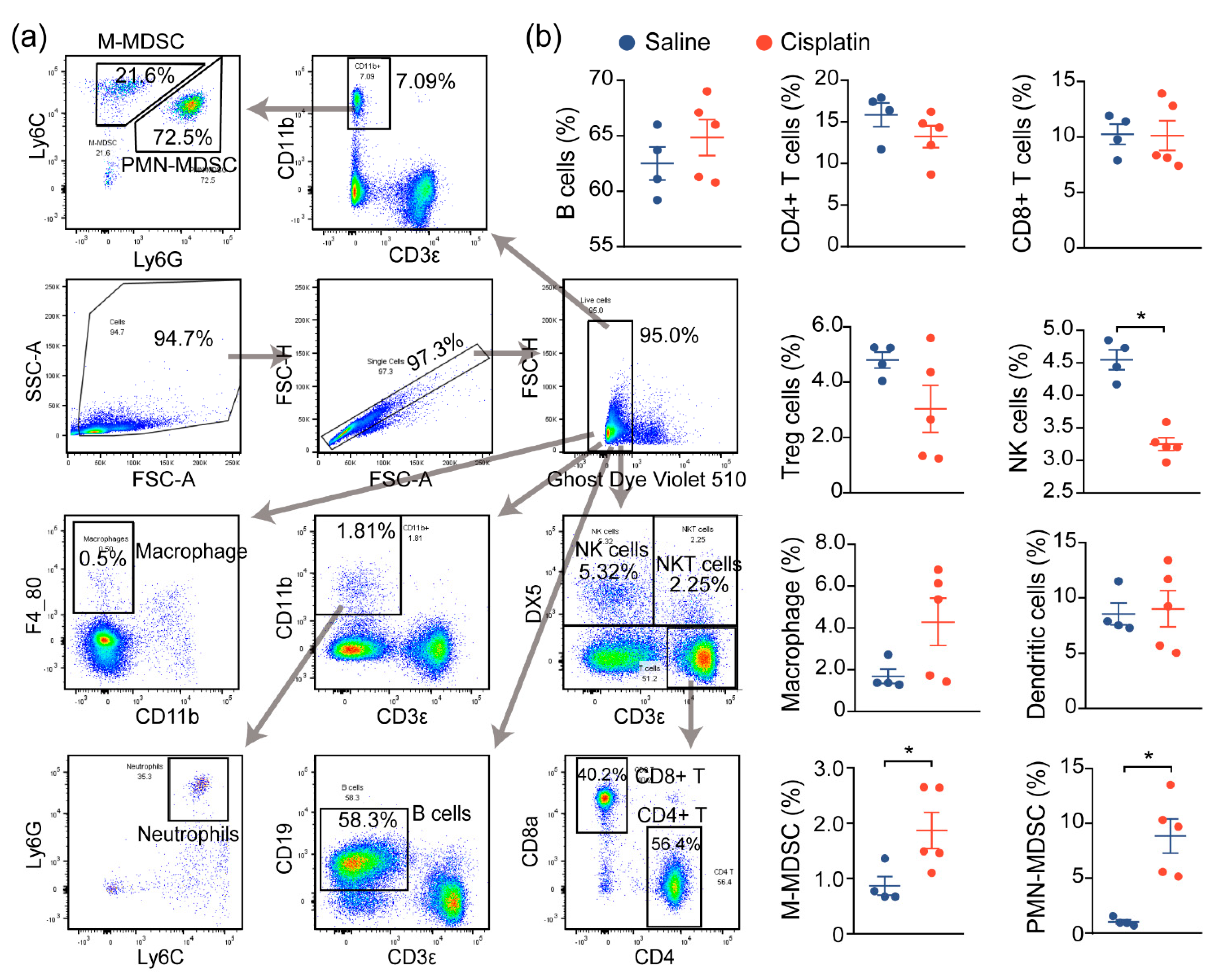
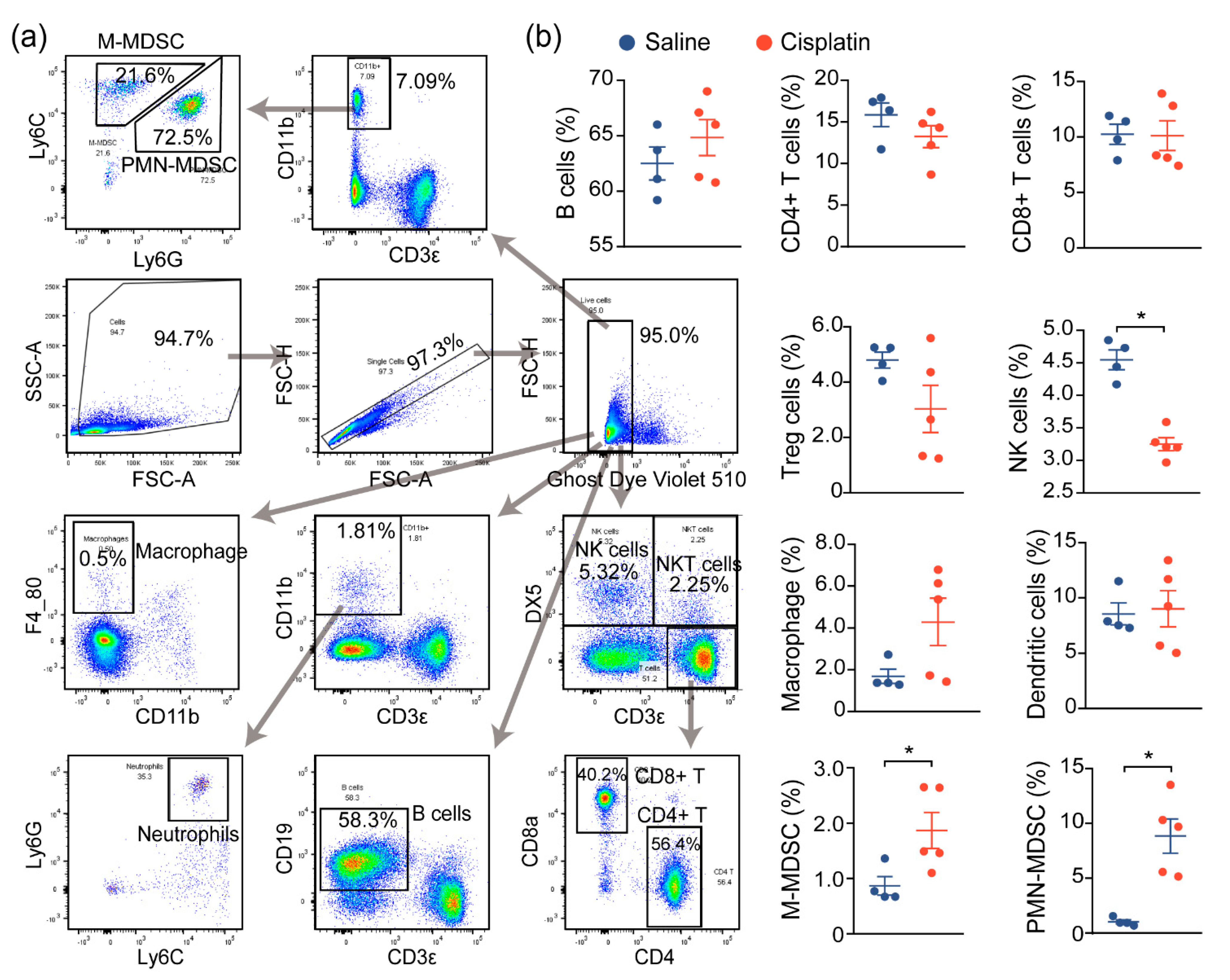
2.2. AKI Mice Exhibit an Imbalanced Immune Response
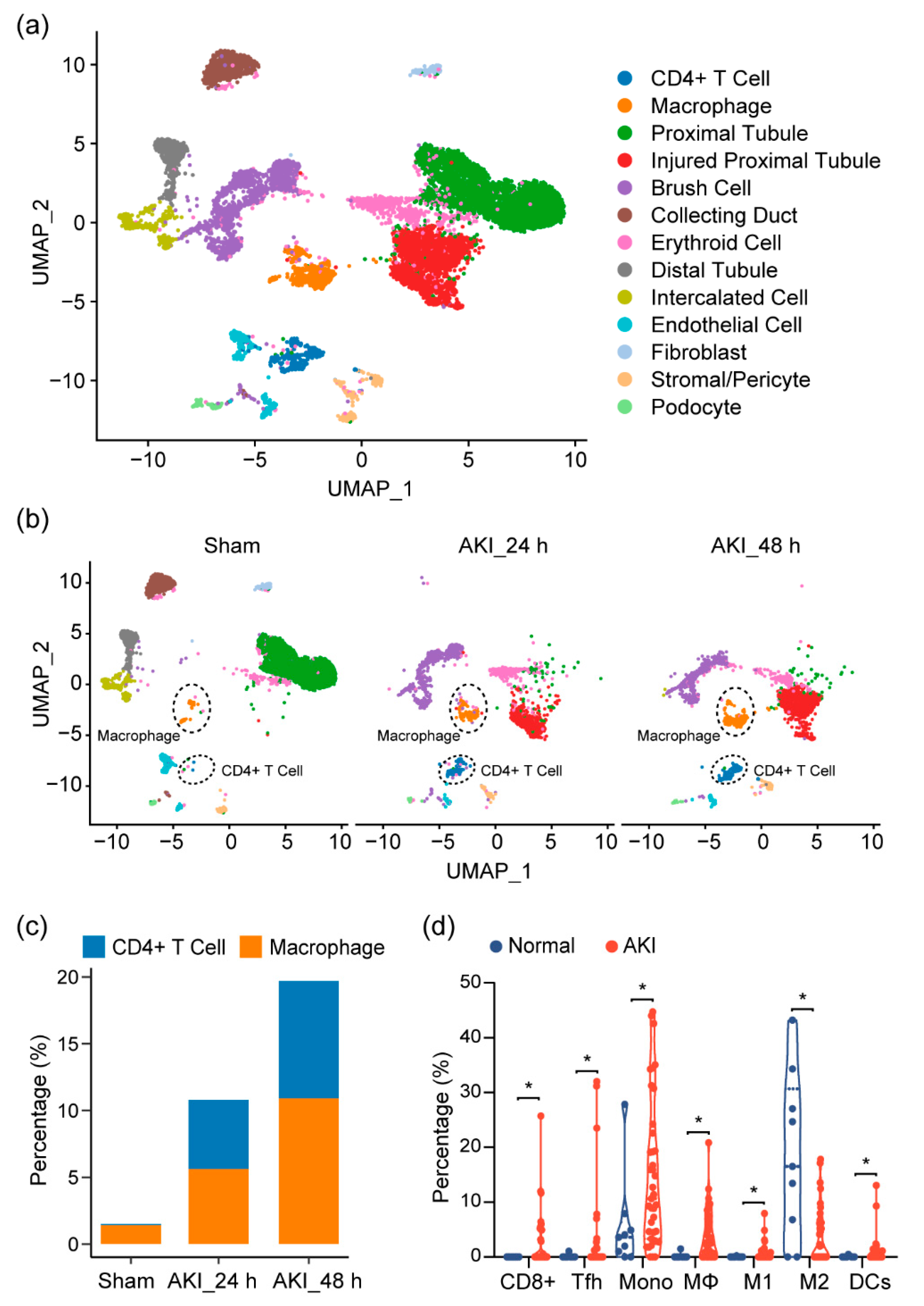
2.3. Immune Characterization and Profiles of AKI
2.4. PD-L1 Dysregulation in Renal Epithelium upon Kidney Injury
2.5. Targeting PD-L1 Protects against AKI
3. Discussion
3.1. Activated Immune System during AKI
3.2. Immune Changes in the Kidney
3.3. T-Cell Changes in AKI
3.4. The PD-1/PD-L1 Axis in Cisplatin-Induced AKI
3.5. Concluding Remarks
4. Materials and Methods
4.1. Mouse Model of Cisplatin-Induced AKI and PD-L1 Orthotopic Expression
4.2. Hematoxylin and Eosin (H&E) Staining Measurement
4.3. Immunohistochemistry
4.4. Cell Culture
4.5. Western Blot Analysis
4.6. Flow Cytometry Assays
4.7. Single-Cell mRNA Sequencing (scRNA-seq) Data Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Moore, B.J.; Torio, C.M. Acute Renal Failure Hospitalizations, 2005-2014: Statistical Brief #231. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2006. [Google Scholar]
- Schetz, M.; Dasta, J.; Goldstein, S.; Golper, T. Drug-induced acute kidney injury. Curr. Opin. Crit. Care 2005, 11, 555–565. [Google Scholar] [CrossRef]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. Ther. 2019, 11, 97. [Google Scholar] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Formeck, C.L.; Kellum, J.A. Kidney-Immune System Crosstalk in AKI. Semin. Nephrol. 2019, 39, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2018, 33, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Späth, M.R.; Bartram, M.P.; Palacio-Escat, N.; Hoyer, K.J.R.; Debes, C.; Demir, F.; Schroeter, C.B.; Mandel, A.M.; Grundmann, F.; Ciarimboli, G.; et al. The proteome microenvironment determines the protective effect of preconditioning in cisplatin-induced acute kidney injury. Kidney Int. 2019, 95, 333–349. [Google Scholar] [CrossRef]
- Stam, S.P.; Eisenga, M.F.; Gomes-Neto, A.W.; van Londen, M.; de Meijer, V.E.; van Beek, A.P.; Gansevoort, R.T.; Bakker, S.J.L. Muscle mass determined from urinary creatinine excretion rate, and muscle performance in renal transplant recipients. J. Cachexia Sarcopenia Muscle 2019, 10, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.F.; Cai, R.M.; Lin, Q.; Ye, Q.J.; Ren, J.H.; Yin, L.H.; Li, X. Expansion of polymorphonuclear myeloid-derived suppressor cells in patients with end-stage renal disease may lead to infectious complications. Kidney Int. 2017, 91, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Han, H.I.; Skvarca, L.B.; Espiritu, E.B.; Davidson, A.J.; Hukriede, N.A. The role of macrophages during acute kidney injury: Destruction and repair. Pediatr. Nephrol. 2019, 34, 561–569. [Google Scholar] [CrossRef]
- Gregoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Carrega, P.; Ferlazzo, G. Natural killer cell distribution and trafficking in human tissues. Front Immunol. 2012, 3, 347. [Google Scholar] [CrossRef] [Green Version]
- Tadagavadi, R.K.; Reeves, W.B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Deng, B.; Lin, Y.; Chen, Y.; Ma, S.; Cai, Q.; Wang, W.; Li, B.; Liu, T.; Zhou, P.; He, R.; et al. Plasmacytoid dendritic cells promote acute kidney injury by producing interferon-alpha. Cell Mol Immunol. 2021, 18, 219–229. [Google Scholar] [CrossRef]
- Dellepiane, S.; Leventhal, J.S.; Cravedi, P. T Cells and Acute Kidney Injury: A Two-Way Relationship. Front Immunol. 2020, 11, 1546. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Okusa, M.D. Expanding role of T cells in acute kidney injury. Current Opin. Nephrol. Hypertens. 2014, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chien, C.C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774. [Google Scholar] [CrossRef]
- Nozaki, Y.; Nikolic-Paterson, D.J.; Yagita, H.; Akiba, H.; Holdsworth, S.R.; Kitching, A.R. Tim-1 promotes cisplatin nephrotoxicity. Am. J. Physiol. Renal Physiol. 2011, 301, F1098–F1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manohar, S.; Kompotiatis, P.; Thongprayoon, C.; Cheungpasitporn, W.; Herrmann, J.; Herrmann, S.M. Programmed cell death protein 1 inhibitor treatment is associated with acute kidney injury and hypocalcemia: Meta-analysis. Nephrol. Dial. Transplant. 2019, 34, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, F.B.; Kibbelaar, Z.A.; Glezerman, I.G.; Abudayyeh, A.; Mamlouk, O.; Motwani, S.S.; Murakami, N.; Herrmann, S.M.; Manohar, S.; Shirali, A.C.; et al. Clinical Features and Outcomes of Immune Checkpoint Inhibitor-Associated AKI: A Multicenter Study. J. Am. Soc. Nephrol. 2020, 31, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Konkel, J.E.; Frommer, F.; Leech, M.D.; Yagita, H.; Waisman, A.; Anderton, S.M. PD-1 signalling in CD4(+) T cells restrains their clonal expansion to an immunogenic stimulus, but is not critically required for peptide-induced tolerance. Immunology 2010, 130, 92–102. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Lu, D.; Ni, Z.; Liu, X.; Feng, S.; Dong, X.; Shi, X.; Zhai, J.; Mai, S.; Jiang, J.; Wang, Z.; et al. Beyond T Cells: Understanding the Role of PD-1/PD-L1 in Tumor-Associated Macrophages. J. Immunol. Res. 2019, 2019, 1919082. [Google Scholar] [CrossRef] [Green Version]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Segal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Tredaniel, J.; et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [Green Version]
- Zamani, M.R.; Aslani, S.; Salmaninejad, A.; Javan, M.R.; Rezaei, N. PD-1/PD-L and autoimmunity: A growing relationship. Cell Immunol. 2016, 310, 27–41. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Zhang, Y.; Ding, Q.; Wang, S.S.; Rastogi, P.; Dai, D.F.; Lu, D.; Purvis, M.; Cao, C.; Wang, A.; et al. Epithelial innate immunity mediates tubular cell senescence after kidney injury. JCI Insight 2019, 4, e125490. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Statt, S.; Chiu, C.L.; Thai, P.; Arif, M.; Adler, K.B.; Wu, R. Targeting myristoylated alanine-rich C kinase substrate phosphorylation site domain in lung cancer. Mechanisms and therapeutic implications. Am. J. Respir. Crit. Care Med. 2014, 190, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Fong, L.W.; Yu, E.; Wu, R.; Trott, J.F.; Weiss, R.H. Up-regulation of MARCKS in kidney cancer and its potential as a therapeutic target. Oncogene 2017, 36, 3588–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Chang, W.H.; Fong, L.W.; Weiss, R.H.; Yu, S.L.; Chen, C.H. Targeting the insulin-like growth factor-1 receptor in MTAP-deficient renal cell carcinoma. Signal Transduct. Target Ther. 2019, 4, 2. [Google Scholar] [CrossRef]
- Chen, C.H.; Thai, P.; Yoneda, K.; Adler, K.B.; Yang, P.C.; Wu, R. A peptide that inhibits function of Myristoylated Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer metastasis. Oncogene 2014, 33, 3696–3706. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.C.; Li, J.M.; Xu, J.; Oldham, J.; Phan, S.H.; Last, J.A.; Wu, R.; Chen, C.H. Tackling MARCKS-PIP3 circuit attenuates fibroblast activation and fibrosis progression. FASEB J. 2019, 33, 14354–14369. [Google Scholar] [CrossRef] [Green Version]
- Li, J.M.; Yang, D.C.; Oldham, J.; Linderholm, A.; Zhang, J.; Liu, J.; Kenyon, N.J.; Chen, C.H. Therapeutic targeting of argininosuccinate synthase 1 (ASS1)-deficient pulmonary fibrosis. Mol. Ther. 2021, 29, 1487–1500. [Google Scholar] [CrossRef]
- Rudman-Melnick, V.; Adam, M.; Potter, A.; Chokshi, S.M.; Ma, Q.; Drake, K.A.; Schuh, M.P.; Kofron, J.M.; Devarajan, P.; Potter, S.S. Single-Cell Profiling of AKI in a Murine Model Reveals Novel Transcriptional Signatures, Profibrotic Phenotype, and Epithelial-to-Stromal Crosstalk. J. Am. Soc. Nephrol. 2020, 31, 2793–2814. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Yang, D.C.; Zhang, J.; Hsu, S.-W.; Weiss, R.H.; Chen, C.-H. A Novel Renoprotective Strategy: Upregulation of PD-L1 Mitigates Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2021, 22, 13304. https://doi.org/10.3390/ijms222413304
Liu J, Yang DC, Zhang J, Hsu S-W, Weiss RH, Chen C-H. A Novel Renoprotective Strategy: Upregulation of PD-L1 Mitigates Cisplatin-Induced Acute Kidney Injury. International Journal of Molecular Sciences. 2021; 22(24):13304. https://doi.org/10.3390/ijms222413304
Chicago/Turabian StyleLiu, Jun, David C. Yang, Jun Zhang, Ssu-Wei Hsu, Robert H. Weiss, and Ching-Hsien Chen. 2021. "A Novel Renoprotective Strategy: Upregulation of PD-L1 Mitigates Cisplatin-Induced Acute Kidney Injury" International Journal of Molecular Sciences 22, no. 24: 13304. https://doi.org/10.3390/ijms222413304
APA StyleLiu, J., Yang, D. C., Zhang, J., Hsu, S.-W., Weiss, R. H., & Chen, C.-H. (2021). A Novel Renoprotective Strategy: Upregulation of PD-L1 Mitigates Cisplatin-Induced Acute Kidney Injury. International Journal of Molecular Sciences, 22(24), 13304. https://doi.org/10.3390/ijms222413304