
Sequestosome 1 Is Part of the Interaction Network of VAPB



{kind=link}
{kind=link}
{kind=link}
{kind=link}
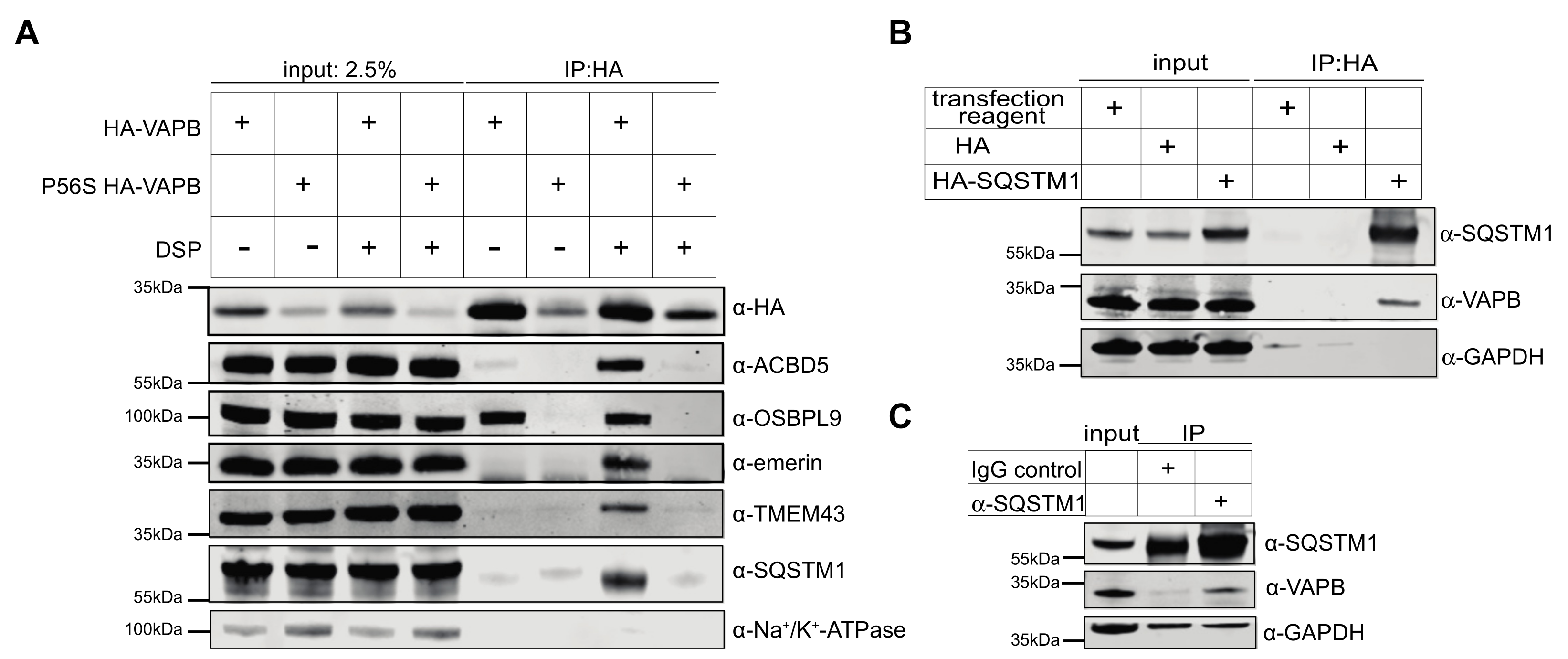{kind=link}
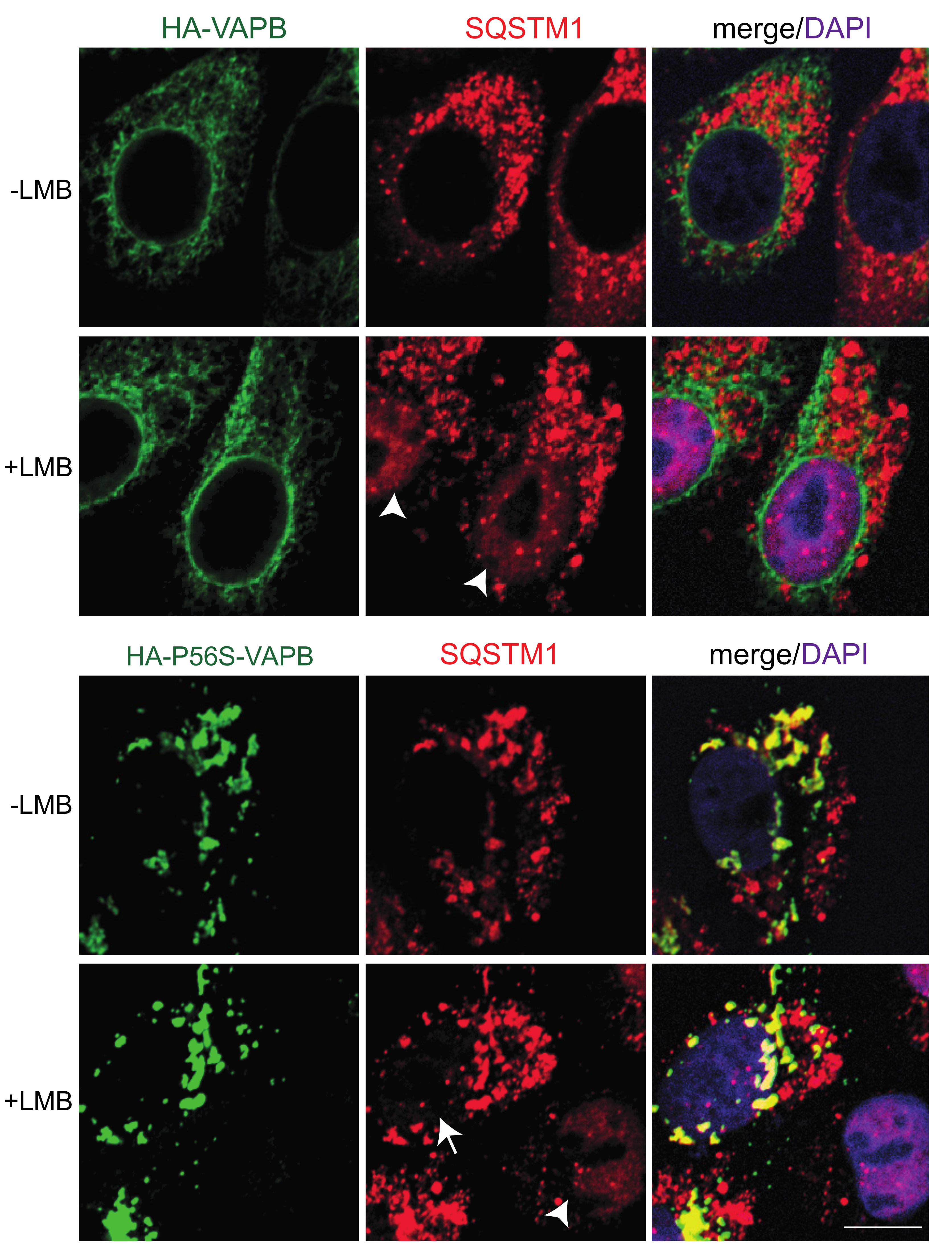{kind=link}
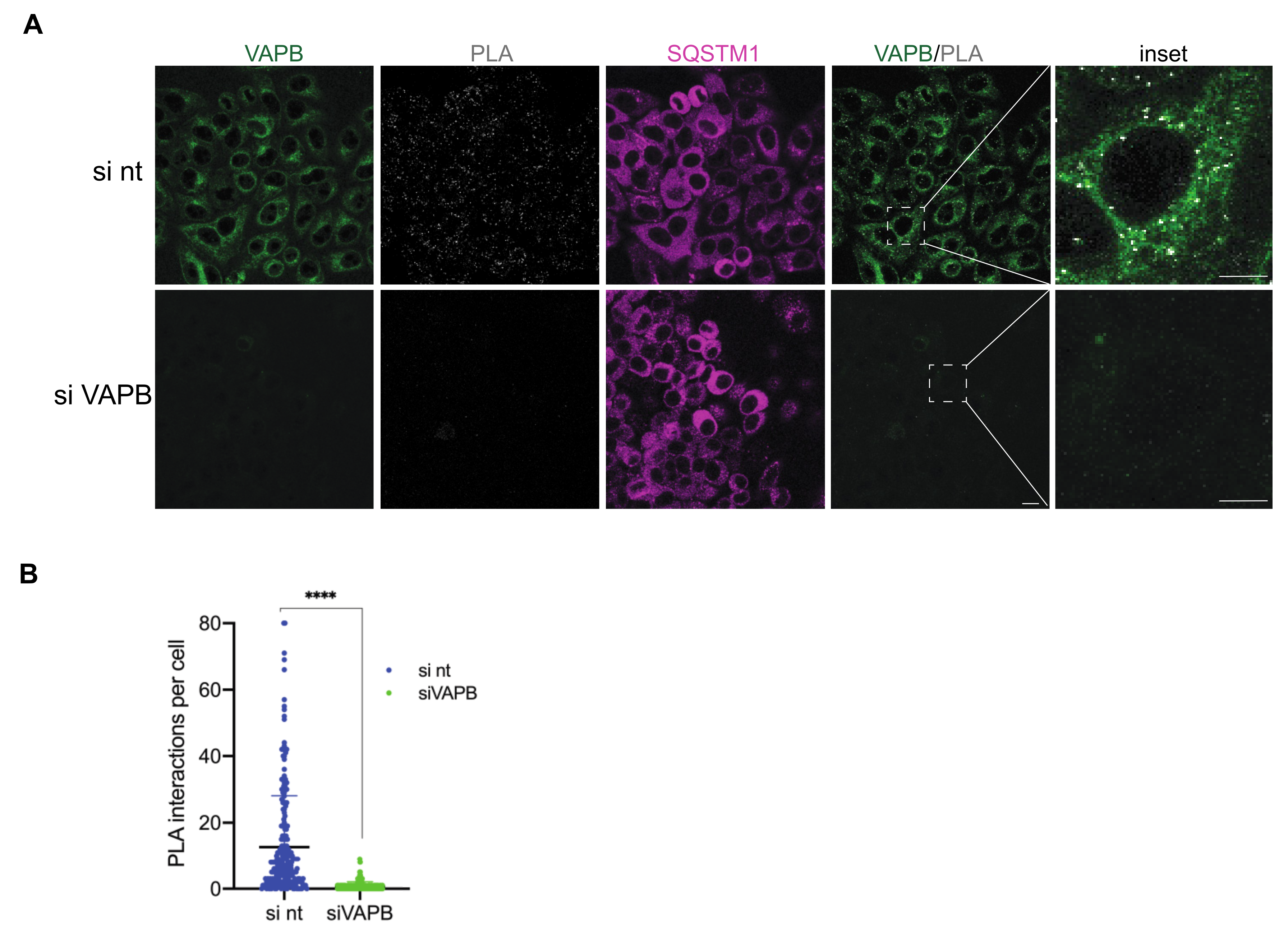{kind=link}
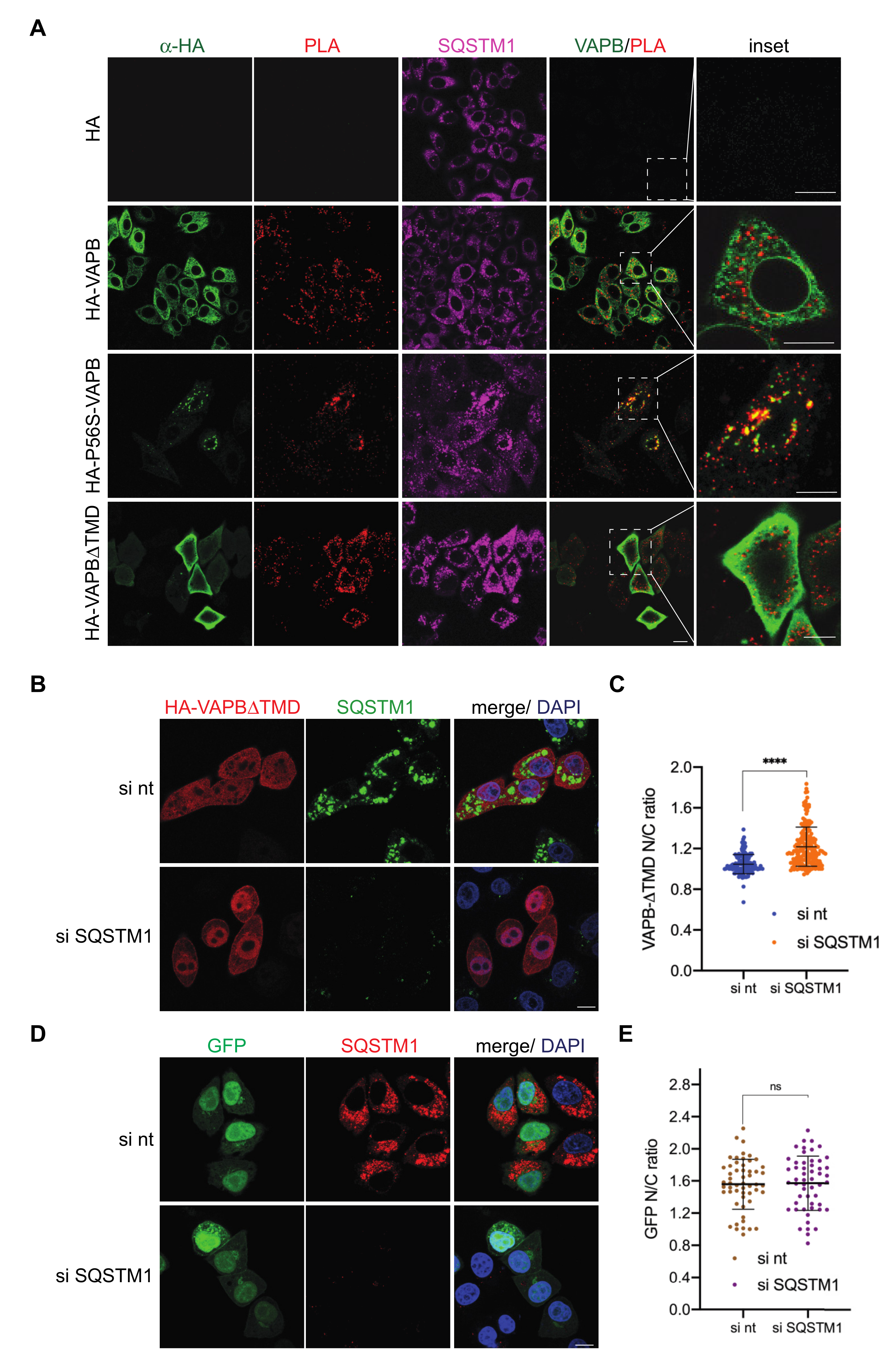{kind=link}
Abstract
:1. Introduction
2. Results
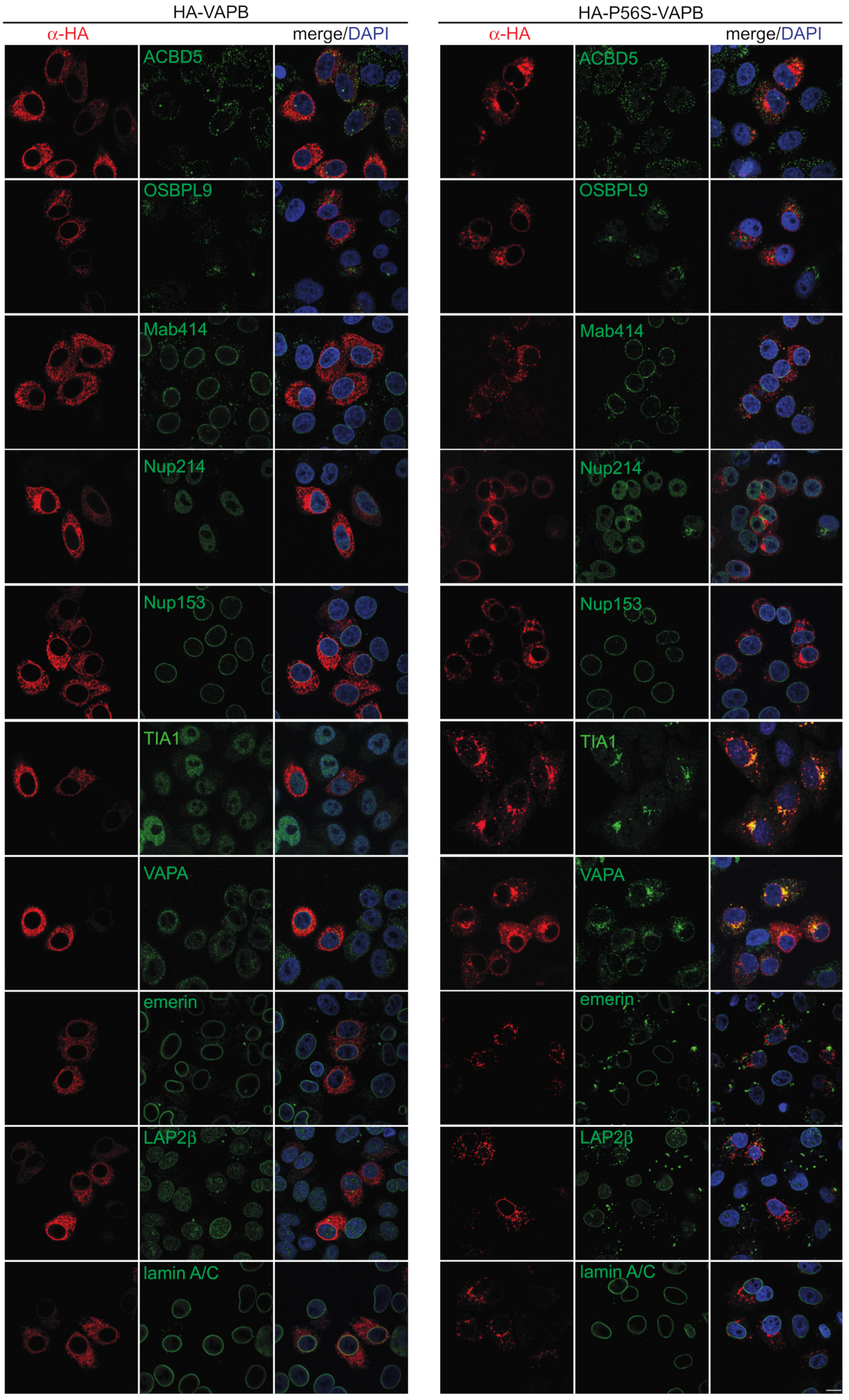
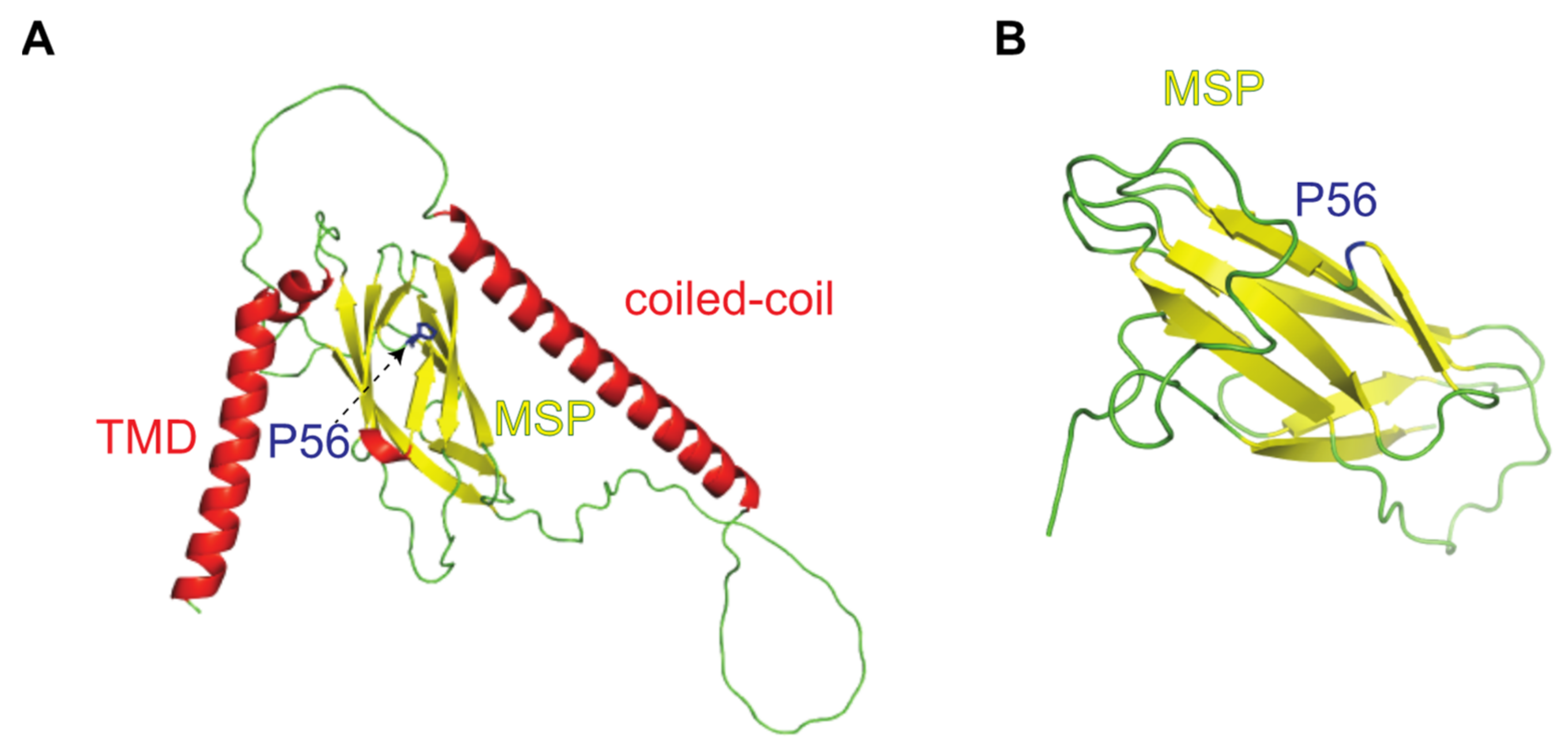
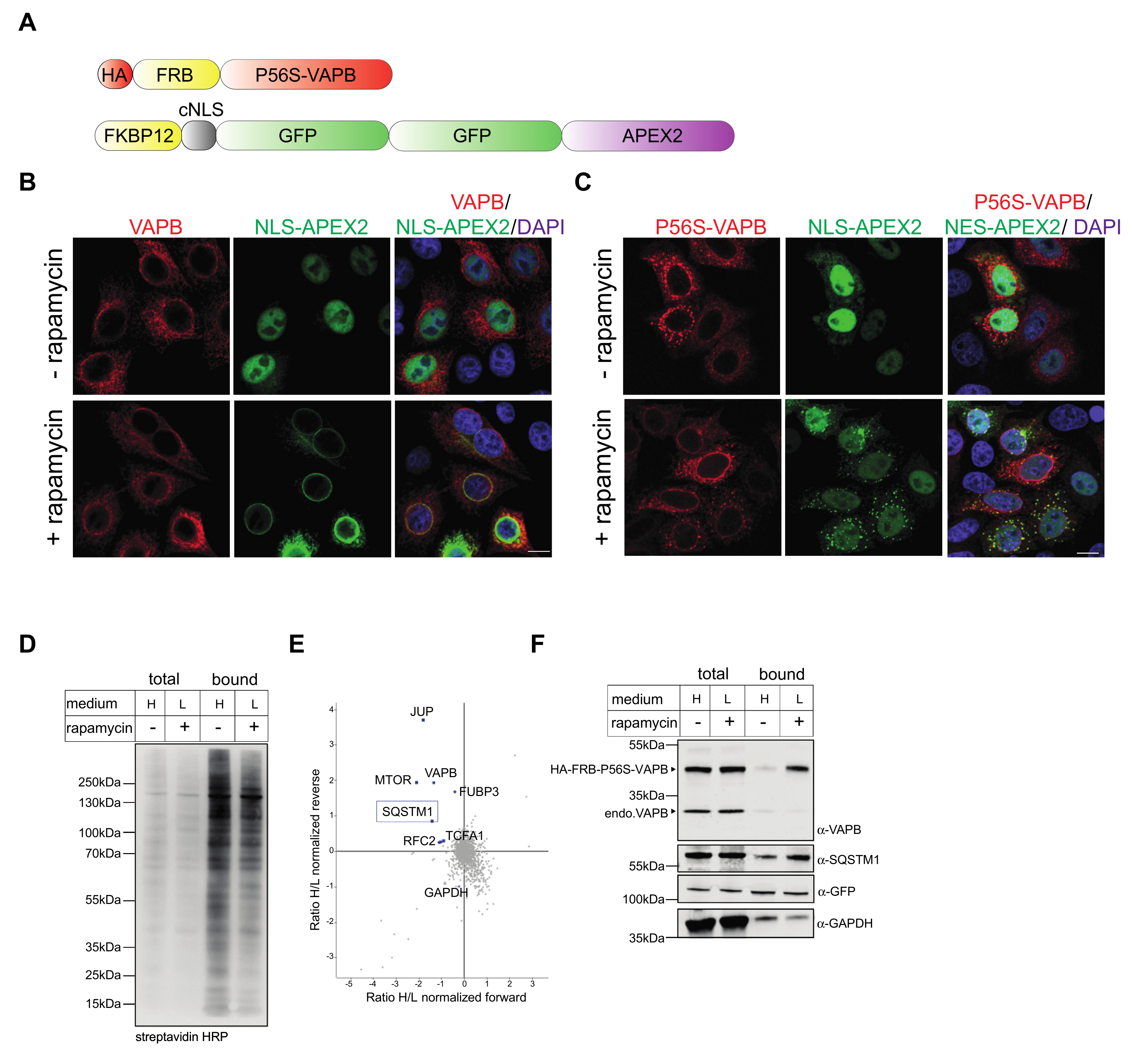
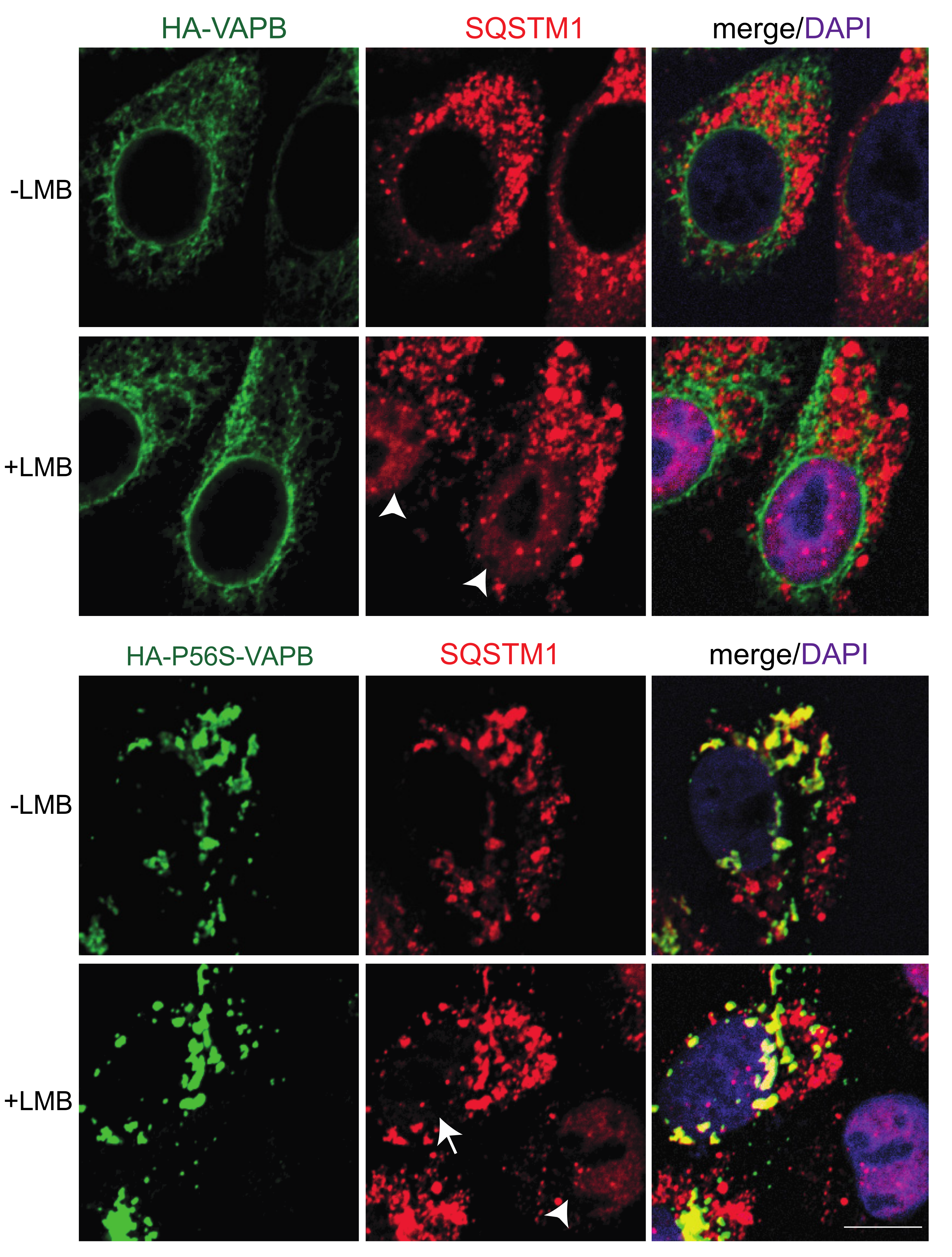
2.1. Effects of P56S-VAPB on Proteins of the Nuclear Envelope
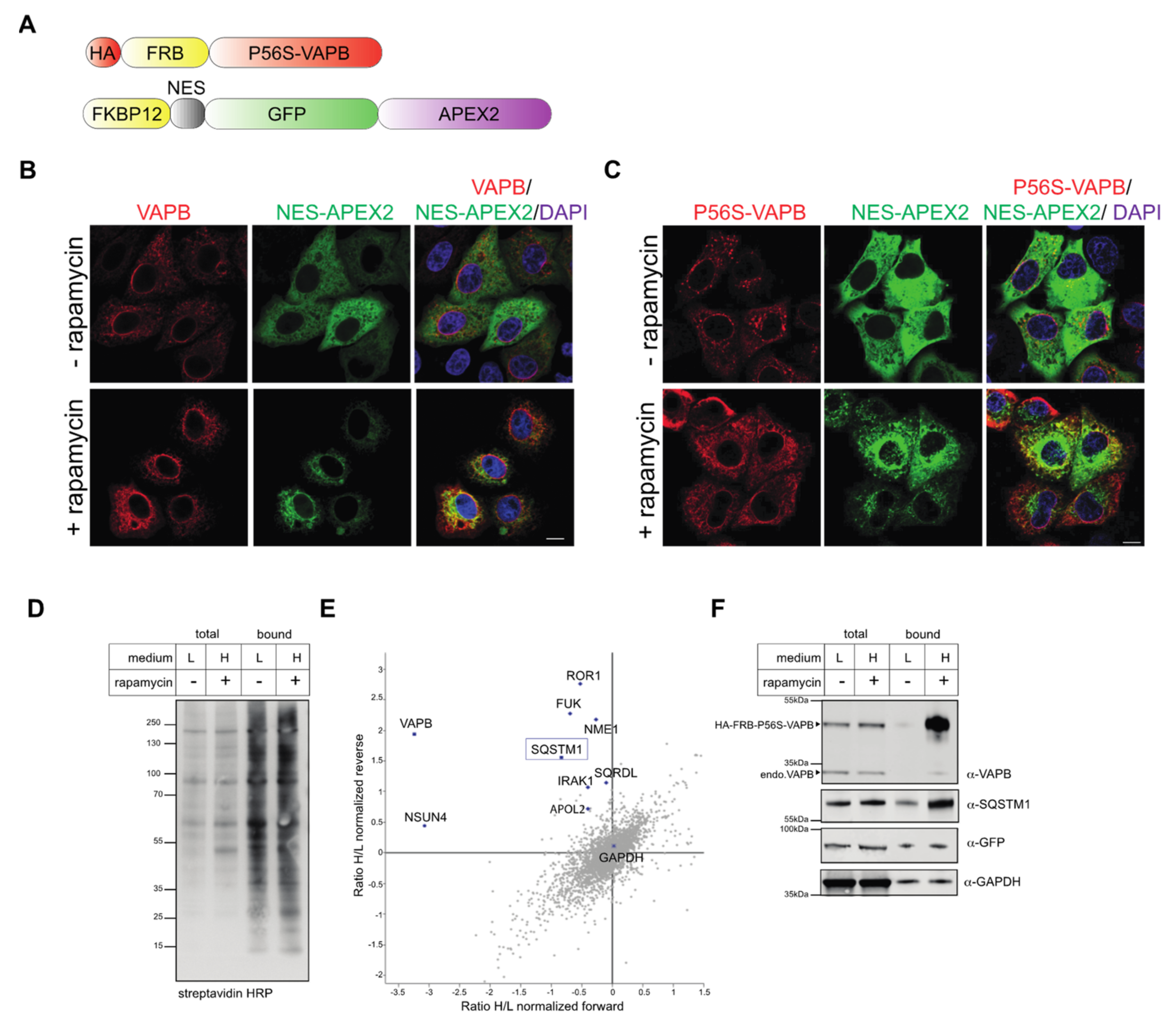
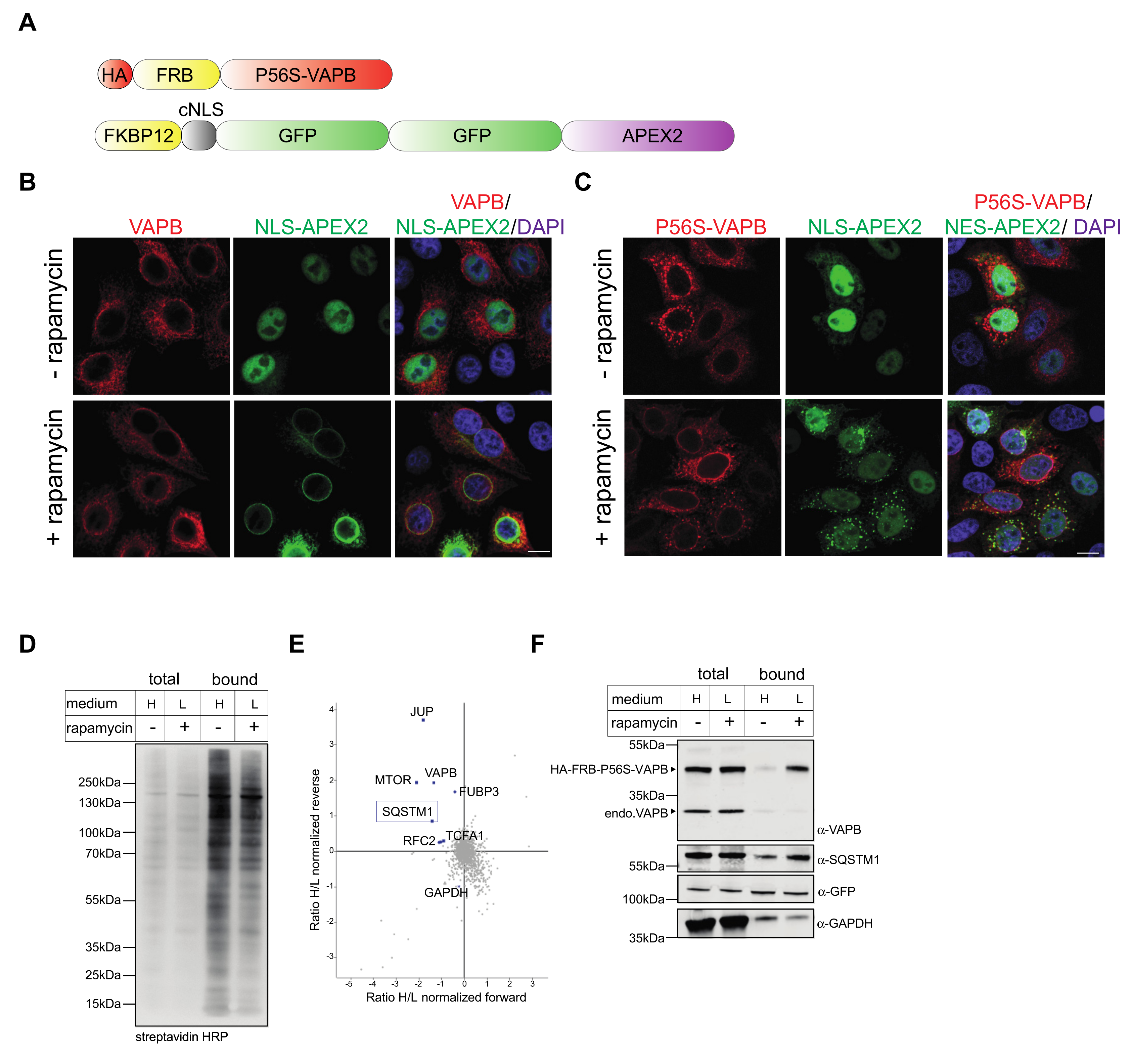
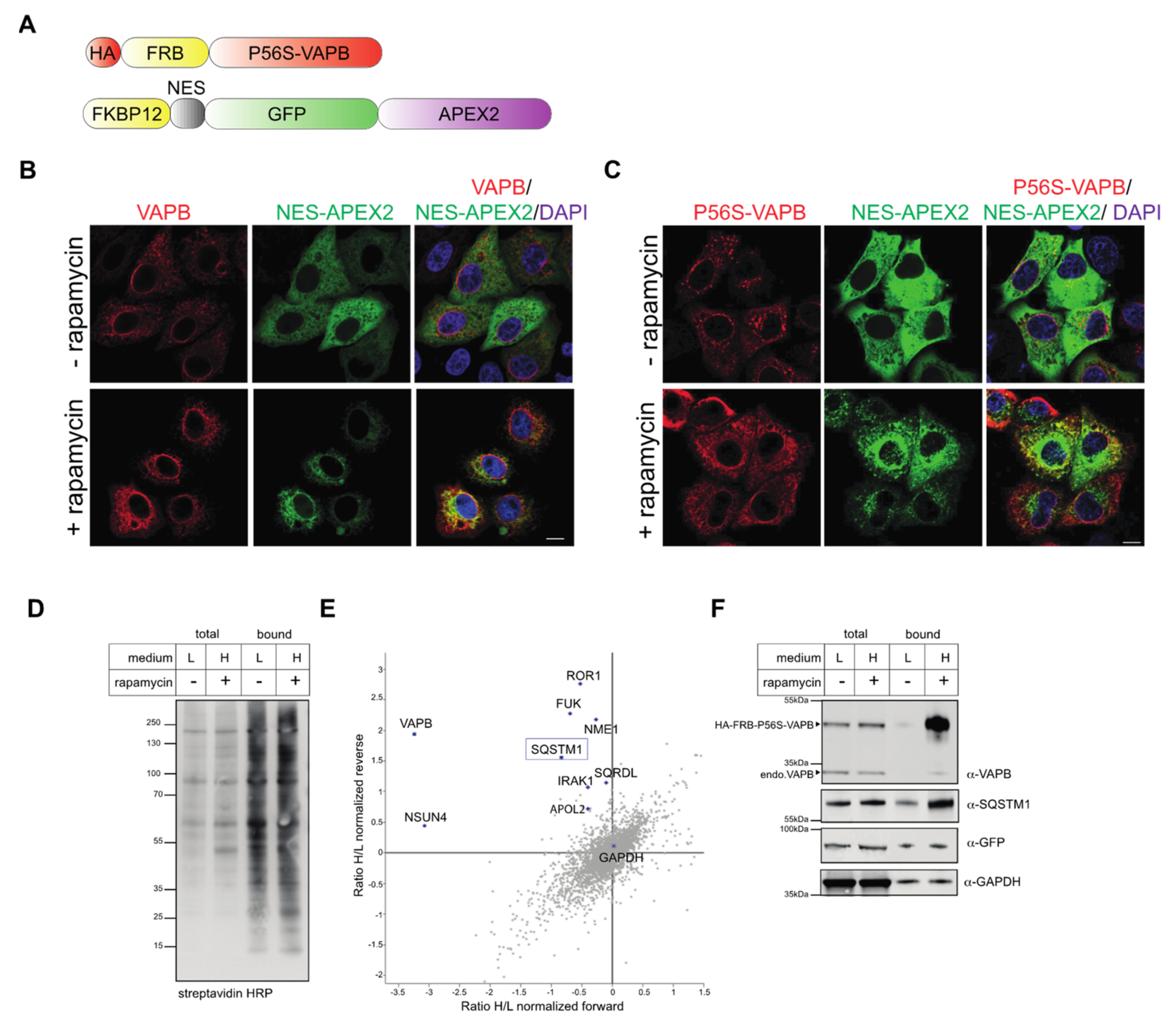
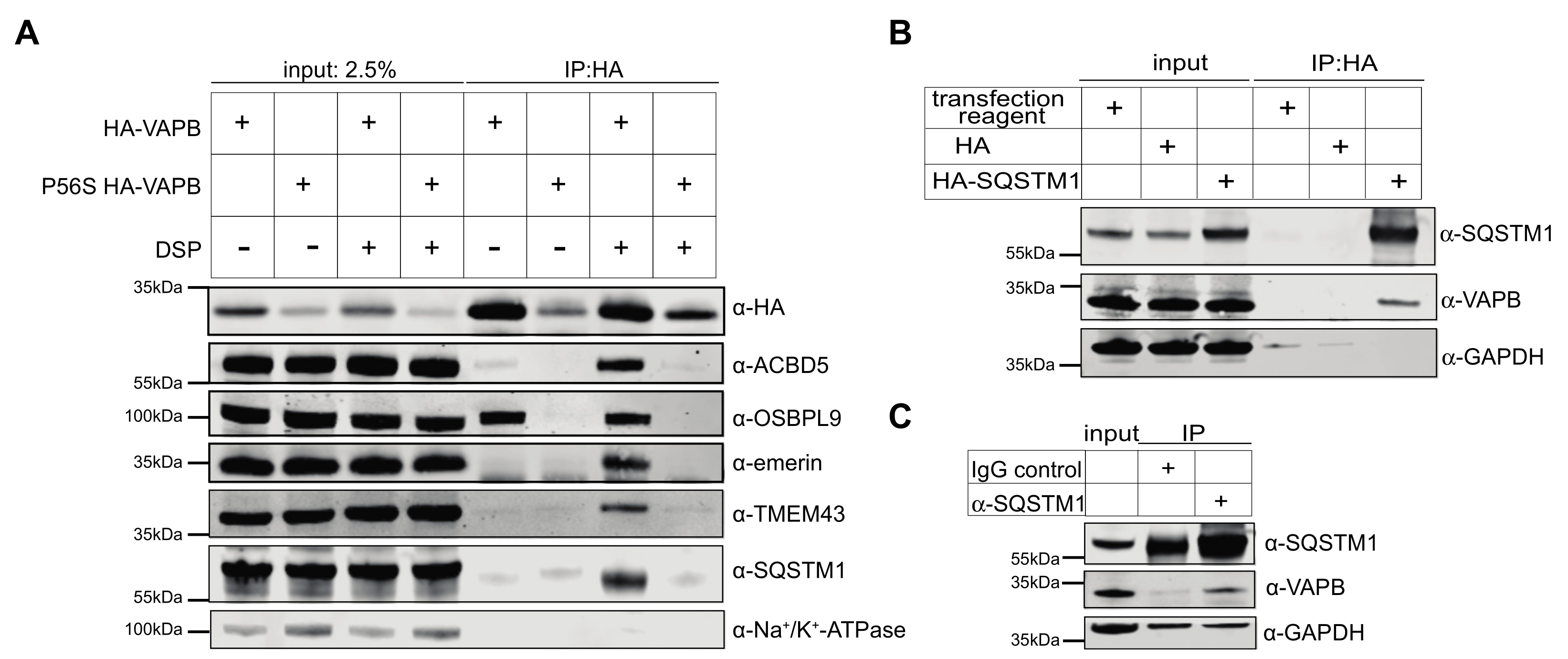
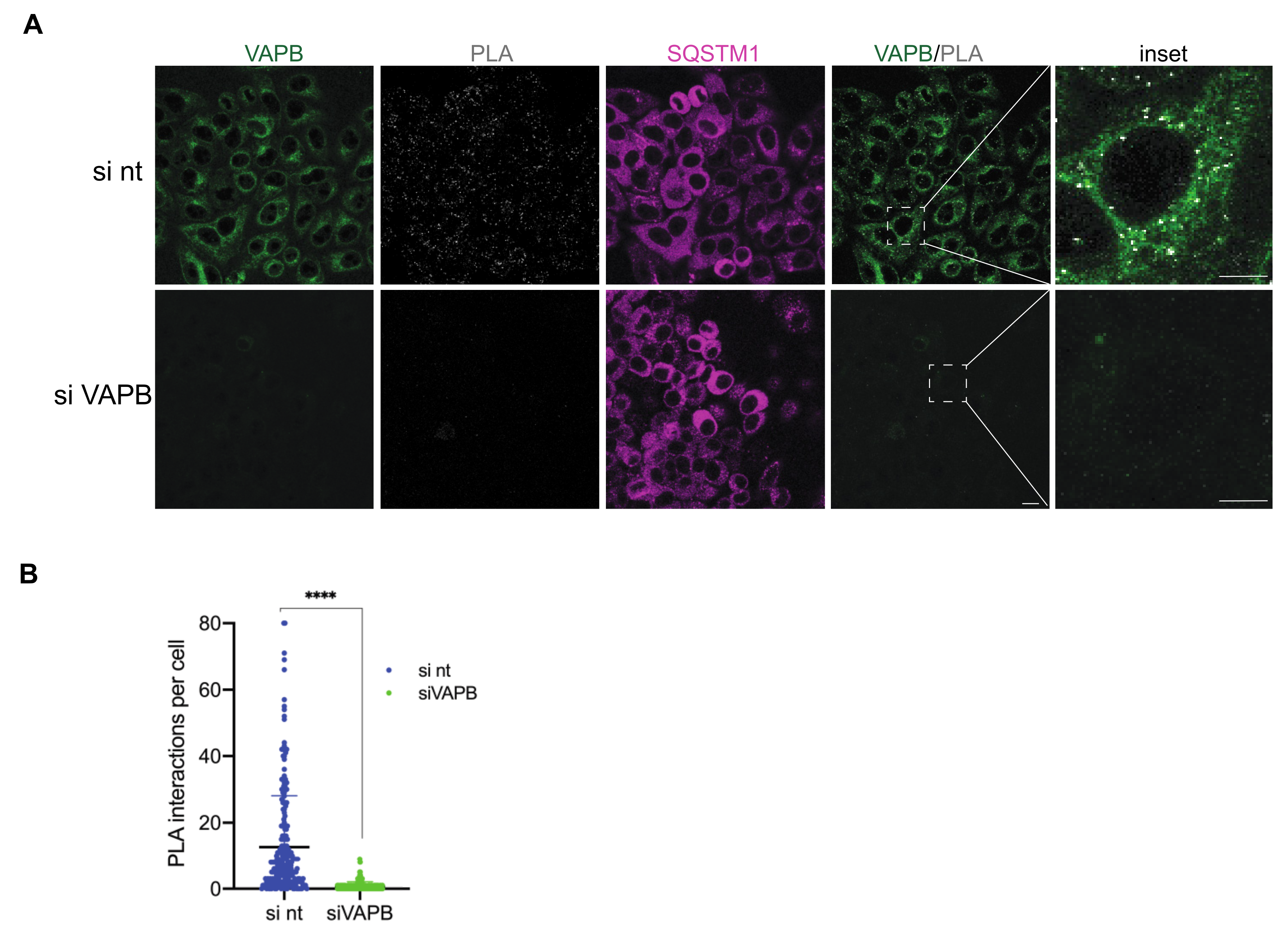
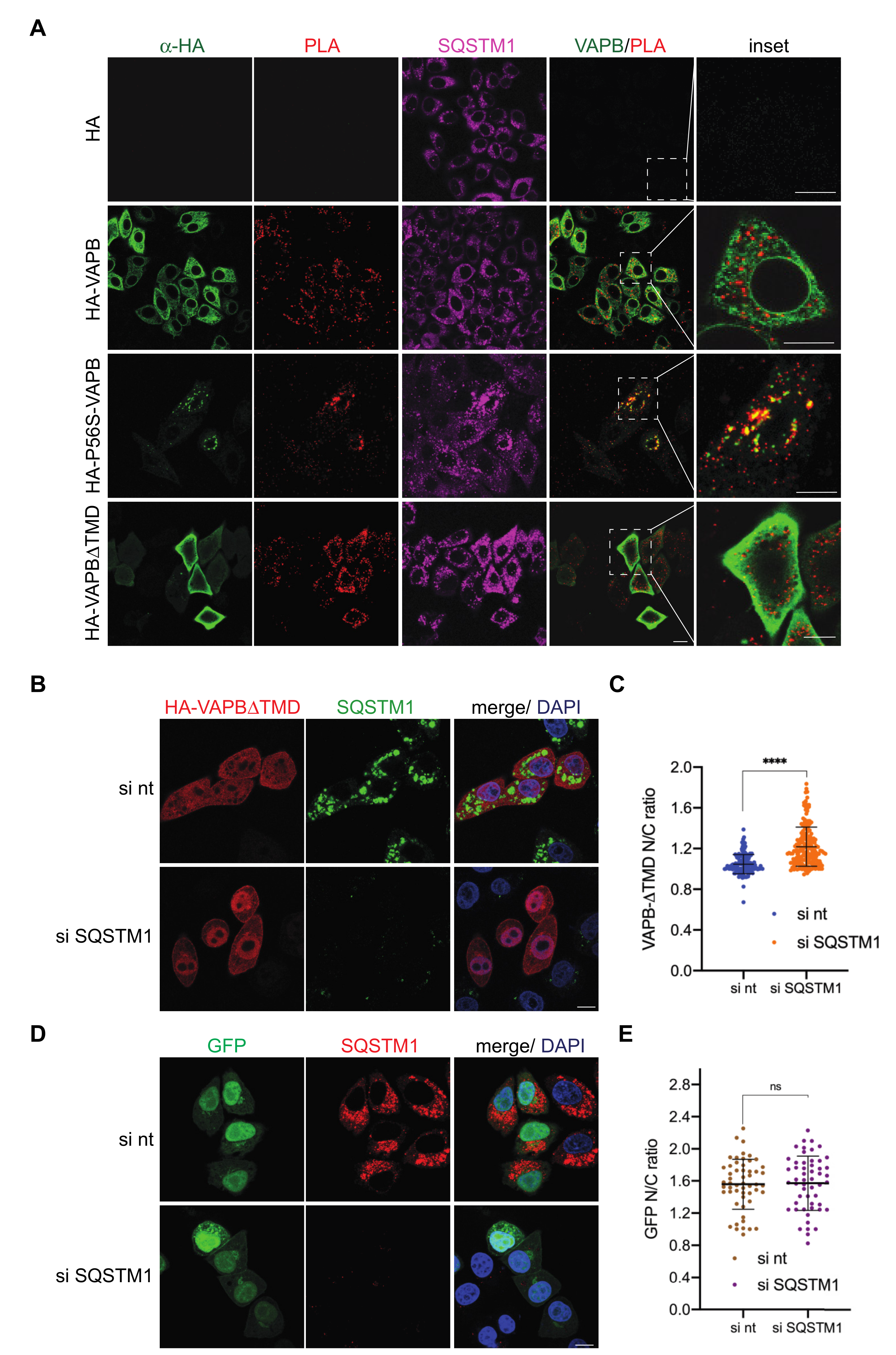
2.2. Identification of Interaction Partners of P56S-VAPB
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Culture and Transfections
4.3. Rapamycin-Dependent Biotinylation
4.4. Mass Spectrometric Analyses
4.5. Data Availability
4.6. Cross-Linking and Coimmunoprecipiation
4.7. Western Blot Analyses
4.8. Immunofluorescence and Fluorescence Microscopy
4.9. Proximity Ligation Assay (PLA)
4.10. Image Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cabukusta, B.; Berlin, I.; van Elsland, D.M.; Forkink, I.; Spits, M.; de Jong, A.W.; Akkermans, J.J.; Wijdeven, R.H.; Janssen, G.M.; van Veelen, P.A.; et al. Human VAPome Analysis Reveals MOSPD1 and MOSPD3 as Membrane Contact Site Proteins Interacting with FFAT-Related FFNT Motifs. Cell Rep. 2020, 33, 108475. [Google Scholar] [CrossRef] [PubMed]
- Di Mattia, T.; Wilhelm, L.P.; Ikhlef, S.; Wendling, C.; Spehner, D.; Nominé, Y.; Giordano, F.; Mathelin, C.; Drin, G.; Tomasetto, C.; et al. Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep. 2018, 19, e45453. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.E.; Brickner, J.H.; Reilein, A.R.; Fenn, T.D.; Walter, P.; Brunger, A.T. Structural Basis of FFAT Motif-Mediated ER Targeting. Structure 2005, 13, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpy, F.; Rousseau, A.; Schwab, Y.; Legueux, F.; Stoll, I.; Wendling, C.; Spiegelhalter, C.; Kessler, P.; Mathelin, C.; Rio, M.-C.; et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci. 2013, 126, 5500–5512. [Google Scholar] [CrossRef] [Green Version]
- Amarilio, R.; Ramachandran, S.; Sabanay, H.; Lev, S. Differential Regulation of Endoplasmic Reticulum Structure through VAP-Nir Protein Interaction. J. Biol. Chem. 2005, 280, 5934–5944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [Green Version]
- Loewen, C.J.R.; Levine, T. A Highly Conserved Binding Site in Vesicle-associated Membrane Protein-associated Protein (VAP) for the FFAT Motif of Lipid-binding Proteins. J. Biol. Chem. 2005, 280, 14097–14104. [Google Scholar] [CrossRef] [Green Version]
- Wyles, J.P.; McMaster, C.R.; Ridgway, N. Vesicle-associated Membrane Protein-associated Protein-A (VAP-A) Interacts with the Oxysterol-binding Protein to Modify Export from the Endoplasmic Reticulum. J. Biol. Chem. 2002, 277, 29908–29918. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Kehlenbach, R. The Interactome of the VAP Family of Proteins: An Overview. Cells 2021, 10, 1780. [Google Scholar] [CrossRef] [PubMed]
- Fasana, E.; Fossati, M.; Ruggiano, A.; Brambillasca, S.; Hoogenraad, C.C.; Navone, F.; Francolini, M.; Borgese, N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010, 24, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Müller, M.; Goldberg, M.W.; Lenz, C.; Urlaub, H.; Kehlenbach, R.H. Proteomic mapping by rapamycin-dependent targeting of APEX2 identifies binding partners of VAPB at the inner nuclear membrane. J. Biol. Chem. 2019, 294, 16241–16254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiz-Ros, N.; Czapiewski, R.; Epifano, I.; Stevenson, A.; Swanson, S.K.; Dixon, C.R.; Zamora, D.B.; McElwee, M.; Vijayakrishnan, S.; Richardson, C.A.; et al. Host Vesicle Fusion Protein VAPB Contributes to the Nuclear Egress Stage of Herpes Simplex Virus Type-1 (HSV-1) Replication. Cells 2019, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Leal, S.S.; Benhalevy, D.; Gomes, C.M.; Lev, S. Structural Requirements for VAP-B Oligomerization and Their Implication in Amyotrophic Lateral Sclerosis-associated VAP-B(P56S) Neurotoxicity. J. Biol. Chem. 2010, 285, 13839–13849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Shi, J.; Lua, S.; Tong, J.S.; Song, J. Elimination of the Native Structure and Solubility of the hVAPB MSP Domain by the Pro56Ser Mutation That Causes Amyotrophic Lateral Sclerosis. Biochemistry 2010, 49, 3887–3897. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040.e28. [Google Scholar] [CrossRef]
- Huttlin, E.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Moustaqim-Barrette, A.; Lin, Y.Q.; Pradhan, S.; Neely, G.G.; Bellen, H.J.; Tsuda, H. The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum. Mol. Genet. 2013, 23, 1975–1989. [Google Scholar] [CrossRef] [Green Version]
- Costello, J.; Castro, I.G.; Hacker, C.; Schrader, T.A.; Metz, J.; Zeuschner, D.; Azadi, A.S.; Godinho, L.F.; Costina, V.; Findeisen, P.; et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 2017, 216, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, M.; Kumagai, K.; Nishijima, M.; Hanada, K. Efficient Trafficking of Ceramide from the Endoplasmic Reticulum to the Golgi Apparatus Requires a VAMP-associated Protein-interacting FFAT Motif of CERT. J. Biol. Chem. 2006, 281, 30279–30288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skehel, P.A.; Martin, K.C.; Kandel, E.R.; Bartsch, D. A VAMP-Binding Protein from Aplysia Required for Neurotransmitter Release. Science 1995, 269, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Soussan, L.; Burakov, D.; Daniels, M.P.; Toister-Achituv, M.; Porat, A.; Yarden, Y.; Elazar, Z. Erg30, a Vap-33–Related Protein, Functions in Protein Transport Mediated by Copi Vesicles. J. Cell Biol. 1999, 146, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Nishimoto, I.; Aiso, S.; Matsuoka, M. Characterization of Amyotrophic Lateral Sclerosis-linked P56S Mutation of Vesicle-associated Membrane Protein-associated Protein B (VAPB/ALS8). J. Biol. Chem. 2006, 281, 30223–30233. [Google Scholar] [CrossRef] [Green Version]
- Kanekura, K.; Suzuki, H.; Aiso, S.; Matsuoka, M. ER Stress and Unfolded Protein Response in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2009, 39, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H., Jr.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, D.; Hatano, M.; Suzuki, N. RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci. Transl. Med. 2017, 9, eaah5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Lim, L.; Wei, Y.; Gupta, G.; Song, J. Resolving the paradox for protein aggregation diseases: NMR structure and dynamics of the membrane-embedded P56S-MSP causing ALS imply a common mechanism for aggregation-prone proteins to attack membranes. F1000Research 2014, 2, 221. [Google Scholar] [CrossRef] [Green Version]
- Papiani, G.; Ruggiano, A.; Fossati, M.; Raimondi, A.; Bertoni, G.; Francolini, M.; Benfante, R.; Navone, F.; Borgese, N. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 2012, 125, 3601–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, H.; Han, S.M.; Yang, Y.; Tong, C.; Lin, Y.Q.; Mohan, K.; Haueter, C.; Zoghbi, A.; Harati, Y.; Kwan, J.; et al. The Amyotrophic Lateral Sclerosis 8 Protein VAPB Is Cleaved, Secreted, and Acts as a Ligand for Eph Receptors. Cell 2008, 133, 963–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgese, N.; Iacomino, N.; Colombo, S.; Navone, F. The Link between VAPB Loss of Function and Amyotrophic Lateral Sclerosis. Cells 2021, 10, 1865. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.; Cheng, D.; Coyaud, E.; Freeman, S.; Di Pietro, E.; Wang, Y.; Vissa, A.; Yip, C.M.; Fairn, G.D.; Braverman, N.; et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 2017, 216, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Silbernagel, N.; Walecki, M.; Schäfer, M.K.; Kessler, M.; Zobeiri, M.; Rinné, S.; Kiper, A.K.; Komadowski, M.A.; Vowinkel, K.S.; Wemhöner, K.; et al. The VAMP-associated protein VAPB is required for cardiac and neuronal pacemaker channel function. FASEB J. 2018, 32, 6159–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijpers, M.; Yu, K.L.; Teuling, E.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. The ALS8 protein VAPB interacts with the ER—Golgi recycling protein YIF1A and regulates membrane delivery into dendrites. EMBO J. 2013, 32, 2056–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, P.; Guo, H.; Dreser, A.; Yamoah, A.; Sechi, A.; Jesse, C.M.; Katona, I.; Doukas, P.; Nikolin, S.; Ernst, S.; et al. Pathomechanisms of ALS8: Altered autophagy and defective RNA binding protein (RBP) homeostasis due to the VAPB P56S mutation. Cell Death Dis. 2021, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Chalhoub, A.; Schooley, A.; Zhang, W.; Ngsee, J.K. A mutation in VAPB that causes amyotrophic lateral sclerosis also causes a nuclear envelope defect. J. Cell Sci. 2012, 125, 2831–2836. [Google Scholar] [CrossRef] [Green Version]
- Teuling, E.; Ahmed, S.; Haasdijk, E.; Demmers, J.; Steinmetz, M.O.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. Motor Neuron Disease-Associated Mutant Vesicle-Associated Membrane Protein-Associated Protein (VAP) B Recruits Wild-Type VAPs into Endoplasmic Reticulum-Derived Tubular Aggregates. J. Neurosci. 2007, 27, 9801–9815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; James, C.; Lenz, C.; Urlaub, H.; Kehlenbach, R.H. Probing the Environment of Emerin by Enhanced Ascorbate Peroxidase 2 (APEX2)-Mediated Proximity Labeling. Cells 2020, 9, 605. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zheng, X.F.; Brown, E.J.; Schreiber, S.L. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl. Acad. Sci. USA 1995, 92, 4947–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobingier, B.T.; Hüttenhain, R.; Eichel, K.; Miller, K.B.; Ting, A.Y.; von Zastrow, M.; Krogan, N.J. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 2017, 169, 350–360.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, I.; Strominger, J.L.; Shin, J. Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc. Natl. Acad. Sci. USA 1996, 93, 5991–5995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadlamudi, R.K.; Joung, I.; Strominger, J.L.; Shin, J. p62, a Phosphotyrosine-independent Ligand of the SH2 Domain of p56lck, Belongs to a New Class of Ubiquitin-binding Proteins. J. Biol. Chem. 1996, 271, 20235–20237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakar, K.; Karaca, S.; Port, S.A.; Urlaub, H.; Kehlenbach, R.H. Identification of CRM1-dependent Nuclear Export Cargos Using Quantitative Mass Spectrometry. Mol. Cell. Proteom. 2013, 12, 664–678. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Lamark, T.; Bruun, J.-A.; Overvatn, A.; Bjorkoy, G.; Johansen, T. Nucleocytoplasmic Shuttling of p62/SQSTM1 and Its Role in Recruitment of Nuclear Polyubiquitinated Proteins to Promyelocytic Leukemia Bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstrale, K.; Leuchowius, K.-J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.-G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Chattopadhyay, D.; Sengupta, S. First evidence of pathogenicity of V234I mutation of hVAPB found in Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2014, 448, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; de Belleroche, J.S. Characterization of the Properties of a Novel Mutation in VAPB in Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2010, 285, 40266–40281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Blitterswijk, M.; van Es, M.; Koppers, M.; van Rheenen, W.; Medic, J.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging 2012, 33, 2950.e1–2950.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Nishiyama, R.; Shimogori, T.; Nukina, N. Proteomics-Based Approach Identifies Altered ER Domain Properties by ALS-Linked VAPB Mutation. Sci. Rep. 2020, 10, 7610. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillée, S.; Doukouré, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. p62/SQSTM 1: ‘Jack of all trades’ in health and cancer. FEBS J. 2018, 286, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larroquette, F.; Seto, L.; Gaub, P.L.; Kamal, B.; Wallis, D.; Larivière, R.; Vallée, J.; Robitaille, R.; Tsuda, H. Vapb/Amyotrophic lateral sclerosis 8 knock-in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response. Hum. Mol. Genet. 2015, 24, 6515–6529. [Google Scholar] [CrossRef] [Green Version]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.-Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol. Cell 2019, 74, 330–346.e11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Liu, N.; Miao, G.; Chen, Y.; Zhao, H.; Zhang, H. The ER Contact Proteins VAPA/B Interact with Multiple Autophagy Proteins to Modulate Autophagosome Biogenesis. Curr. Biol. 2018, 28, 1234–1245.e4. [Google Scholar] [CrossRef]
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Berkamp, S.; Mostafavi, S.; Sachse, C. Structure and function of p62/SQSTM1 in the emerging framework of phase separation. FEBS J. 2020, 10, 1111. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Hao, Z.; Ren, H.; Wang, G. Loss of VAPB Regulates Autophagy in a Beclin 1-Dependent Manner. Neurosci. Bull. 2018, 34, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, J.; Monroy, J.R.; Jamieson, C.; Rajanala, K.; Vilardi, F.; Schwappach, B.; Kehlenbach, R.H. Emery-Dreifuss muscular dystrophy mutations impair TRC40-mediated targeting of emerin to the inner nuclear membrane. J. Cell Sci. 2016, 129, 502–516. [Google Scholar] [CrossRef] [Green Version]
- Charneau, P.; Mirambeau, G.; Roux, P.; Paulous, S.; Buc, H.; Clavel, F. HIV-1 Reverse Transcription A Termination Step at the Center of the Genome. J. Mol. Biol. 1994, 241, 651–662. [Google Scholar] [CrossRef]
- Chen, C.; Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell Biol. 1987, 7, 2745–2752. [Google Scholar]
- Atanassov, I.; Urlaub, H. Increased proteome coverage by combining PAGE and peptide isoelectric focusing: Comparative study of gel-based separation approaches. Proteomics 2013, 13, 2947–2955. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.-A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, C.; Lenz, C.; Urlaub, H.; Kehlenbach, R.H. Sequestosome 1 Is Part of the Interaction Network of VAPB. Int. J. Mol. Sci. 2021, 22, 13271. https://doi.org/10.3390/ijms222413271
James C, Lenz C, Urlaub H, Kehlenbach RH. Sequestosome 1 Is Part of the Interaction Network of VAPB. International Journal of Molecular Sciences. 2021; 22(24):13271. https://doi.org/10.3390/ijms222413271
Chicago/Turabian StyleJames, Christina, Christof Lenz, Henning Urlaub, and Ralph H. Kehlenbach. 2021. "Sequestosome 1 Is Part of the Interaction Network of VAPB" International Journal of Molecular Sciences 22, no. 24: 13271. https://doi.org/10.3390/ijms222413271
APA StyleJames, C., Lenz, C., Urlaub, H., & Kehlenbach, R. H. (2021). Sequestosome 1 Is Part of the Interaction Network of VAPB. International Journal of Molecular Sciences, 22(24), 13271. https://doi.org/10.3390/ijms222413271