
The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis


Abstract
1. Introduction
2. Genetic Causes of ALS
2.1. Superoxide Dismutase 1
2.2. TAR DNA Binding Protein 43
2.3. Fused in Sarcoma
2.4. Chromosome 9 Open Reading Frame 72
3. ALS Histopathology
4. Nucleocytoplasmic Transport
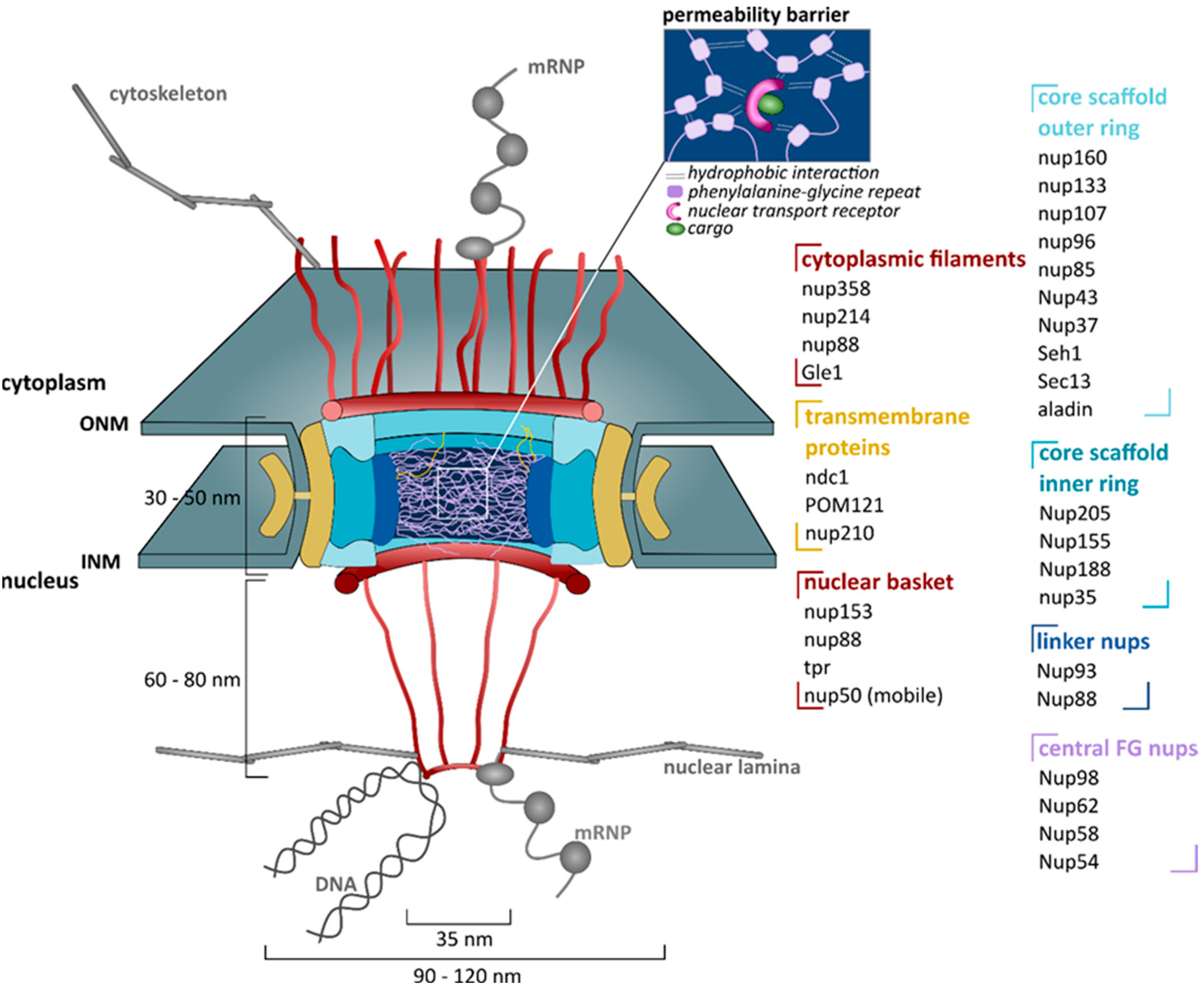
4.1. The Nuclear Pore Complex (NPC)
4.1.1. Structure
4.1.2. Functions
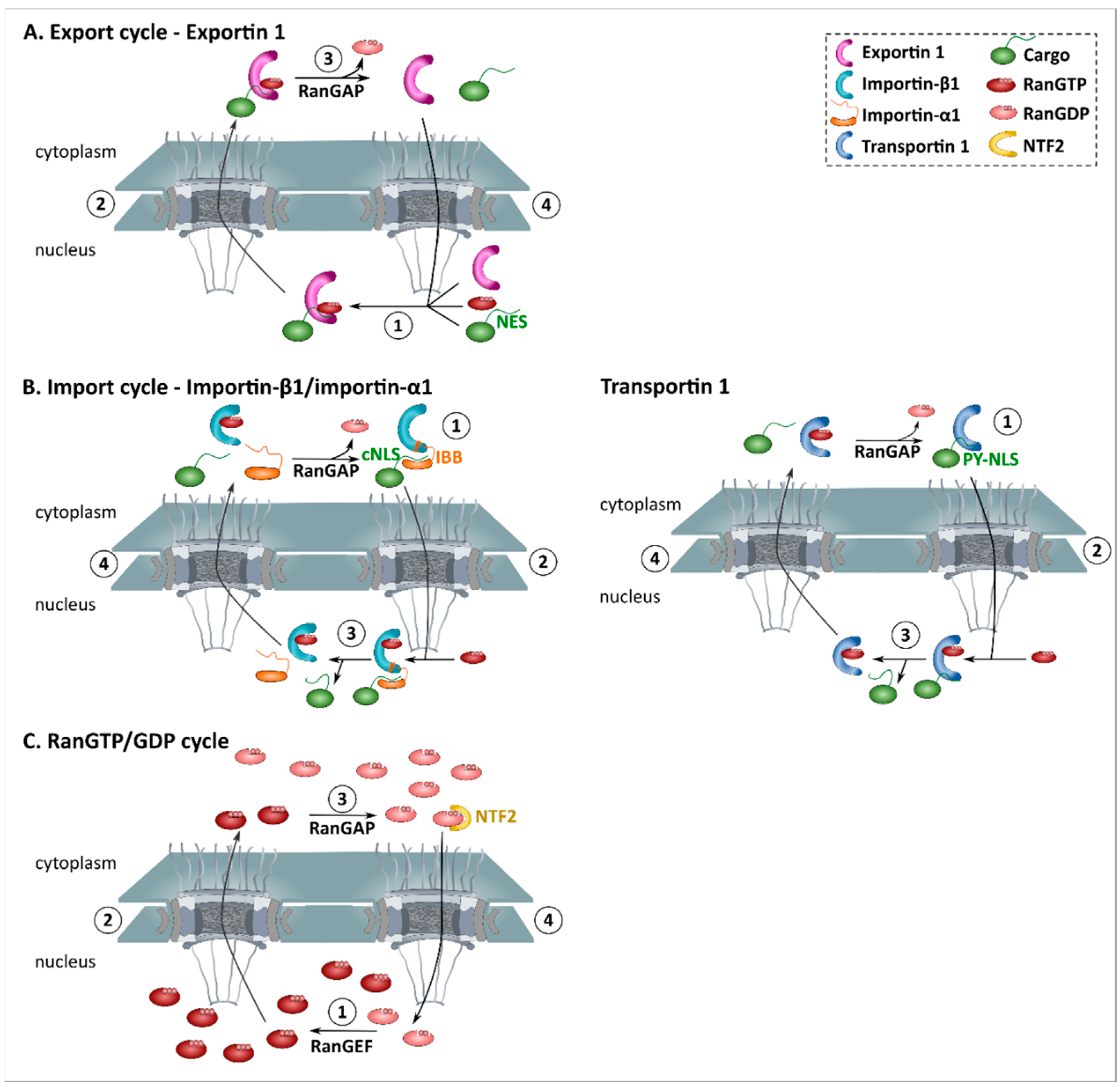
4.2. Nuclear Transport Receptors (NTRs)
4.2.1. Introduction
4.2.2. Functions
4.3. The RanGTP Gradient
4.3.1. Introduction
4.3.2. Function
5. Evidence for Defective Nucleocytoplasmic Transport in ALS
5.1. Aberrant Subcellular Localization of Proteins
5.1.1. Mislocalization of Nucleocytoplasmic Transport Proteins
5.1.2. Mislocalization of Cargoes
5.2. Nucleocytoplasmic Transport Proteins as Modifiers of Disease
5.3. Functional Assays
6. Therapeutic Potential
7. Nucleocytoplasmic Transport and the Aging Brain
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharm. 2017, 18, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Ghasemi, M.; Brown, R.H. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 2548. [Google Scholar] [CrossRef][Green Version]
- Bogaert, E.; Boeynaems, S.; Kato, M.; Guo, L.; Caulfield, T.R.; Steyaert, J.; Scheveneels, W.; Wilmans, N.; Haeck, W.; Hersmus, N.; et al. Molecular dissection of FUS points at synergistic effect of low-complexity domains in toxicity. Cell Rep. 2018, 24, 529–537. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Van der Zee, J.; Gijselinck, I.; Dillen, L.; Van Langenhove, T.; Theuns, J.; Engelborghs, S.; Philtjens, S.; Vandenbulcke, M.; Sleegers, K.; Sieben, A.; et al. A pan-European study of the C9orf72 repeat associated with FTLD: Geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 2013, 34, 363–373. [Google Scholar] [CrossRef]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet. Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef]
- Zhang, D.; Iyer, L.M.; He, F.; Aravind, L. Discovery of Novel DENN proteins: Implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 2012, 3, 283. [Google Scholar] [CrossRef]
- Iyer, S.; Subramanian, V.; Acharya, K.R. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ 2018, 6, e5815. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Campanari, M.-L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; MacNair, L.; McGoldrick, P.; McKeever, P.M.; McLean, J.R.; Zhang, M.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 78, 568–583. [Google Scholar] [CrossRef]
- Frick, P.; Sellier, C.; Mackenzie, I.R.A.; Cheng, C.-Y.; Tahraoui-Bories, J.; Martinat, C.; Pasterkamp, R.J.; Prudlo, J.; Edbauer, D.; Oulad-Abdelghani, M.; et al. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 2018, 6, 72. [Google Scholar] [CrossRef]
- Braems, E.; Swinnen, B.; Van Den Bosch, L. C9orf72 loss-of-function: A trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol. 2020, 140, 625–643. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W.; Van Den Bosch, L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Robin Highley, J.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef]
- Rossi, S.; Serrano, A.; Gerbino, V.; Giorgi, A.; Di Francesco, L.; Nencini, M.; Bozzo, F.; Schininà, M.E.; Bagni, C.; Cestra, G.; et al. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 2015, 128, 1787. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.J.; Shaw, P.G.; Kim, M.-S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef]
- Hartmann, H.; Hornburg, D.; Czuppa, M.; Bader, J.; Michaelsen, M.; Farny, D.; Arzberger, T.; Mann, M.; Meissner, F.; Edbauer, D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 2018, 1, e201800070. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 2016, 167, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef]
- Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.-B. Poly(GR) in C9ORF72-Related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 2017, 65, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, L.; Young, F.L.; Boeynaems, S.; De Decker, M.; Mehta, A.R.; Swijsen, A.; Fazal, R.; Guo, W.; Moisse, M.; Beckers, J.; et al. C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 2021, 7, eabg3013. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PR(n) poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef]
- Hutten, S.; Usluer, S.; Bourgeois, B.; Simonetti, F.; Odeh, H.M.; Fare, C.M.; Czuppa, M.; Hruska-Plochan, M.; Hofweber, M.; Polymenidou, M.; et al. Nuclear import receptors directly bind to arginine-rich dipeptide repeat proteins and suppress their pathological interactions. Cell Rep. 2020, 33, 108538. [Google Scholar] [CrossRef]
- Hayes, L.R.; Duan, L.; Bowen, K.; Kalab, P.; Rothstein, J.D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife 2020, 9, e51685. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Tateishi, T.; Hokonohara, T.; Yamasaki, R.; Miura, S.; Kikuchi, H.; Iwaki, A.; Tashiro, H.; Furuya, H.; Nagara, Y.; Ohyagi, Y.; et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol. 2010, 119, 355–364. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Lanznaster, D.; Hergesheimer, R.; Vourc’h, P.; Corcia, P.; Blasco, H. TDP43 aggregates: The ‘Schrodinger’s cat’ in amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2021, 22, 514. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Reply to ‘TDP43 aggregates: The ‘Schrodinger’s cat’ in amyotrophic lateral sclerosis’. Nat. Rev. Neurosci. 2021, 22, 515. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.A.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.C.; Chook, Y.M. Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc. Natl. Acad. Sci. USA 2012, 109, 12017–12021. [Google Scholar] [CrossRef]
- Kent, L.; Vizard, T.N.; Smith, B.N.; Topp, S.D.; Vance, C.; Gkazi, A.; Miller, J.; Shaw, C.E.; Talbot, K. Autosomal dominant inheritance of rapidly progressive amyotrophic lateral sclerosis due to a truncation mutation in the fused in sarcoma (FUS) gene. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 557–562. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A.; Hetzer, M.W. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 2008, 18, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The structure of the nuclear pore complex (An update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [PubMed]
- Strambio-De-Castillia, C.; Niepel, M.; Rout, M.P. The nuclear pore complex: Bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 490–501. [Google Scholar] [CrossRef]
- Jühlen, R.; Fahrenkrog, B. Moonlighting nuclear pore proteins: Tissue-specific nucleoporin function in health and disease. Histochem. Cell Biol. 2018, 150, 593–605. [Google Scholar] [CrossRef]
- Kim, H.J.; Taylor, J.P. Lost in transportation: Nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.B.; Görlich, D. Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem. Sci. 2016, 41, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Bono, F.; Jinek, M.; Conti, E. Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 2007, 76, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gelles, J.; Musser, S.M. Imaging of single-molecule translocation through nuclear pore complexes. Proc. Natl. Acad. Sci. USA 2004, 101, 12887–12892. [Google Scholar] [CrossRef] [PubMed]
- Ribbeck, K.; Görlich, D. Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 2001, 20, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Ossareh-Nazari, B.; Gwizdek, C.; Dargemont, C. Protein export from the nucleus. Traffic 2001, 2, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Kırlı, K.; Karaca, S.; Dehne, H.J.; Samwer, M.; Pan, K.T.; Lenz, C.; Urlaub, H.; Görlich, D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. eLife 2015, 4, e11466. [Google Scholar] [CrossRef]
- Harel, A.; Forbes, D.J. Importin beta: Conducting a much larger cellular symphony. Mol. Cell 2004, 16, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Görlich, D.; Kutay, U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999, 15, 607–660. [Google Scholar] [CrossRef]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Twyffels, L.; Gueydan, C.; Kruys, V. Transportin-1 and Transportin-2: Protein nuclear import and beyond. FEBS Lett. 2014, 588, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef]
- Cansizoglu, A.E.; Lee, B.J.; Zhang, Z.C.; Fontoura, B.M.A.; Chook, Y.M. Structure-based design of a pathway-specific nuclear import inhibitor. Nat. Struct. Mol. Biol. 2007, 14, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Mingot, J.-M.; Schwarzmaier, P.; Hartmann, E.; Görlich, D. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J. 2002, 21, 377–386. [Google Scholar] [CrossRef]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.-D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 2018, 173, 706–719. [Google Scholar] [CrossRef]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F.; et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 2018, 173, 720–734. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear Import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 2018, 173, 693–705. [Google Scholar] [CrossRef]
- Dasso, M. The ran GTPase: Theme and variations. Curr. Biol. 2002, 12, R502–R508. [Google Scholar] [CrossRef]
- Güttler, T.; Görlich, D. Ran-dependent nuclear export mediators: A structural perspective. EMBO J. 2011, 30, 3457–3474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Yamashita, T.; Kato, H.; Kimura, T.; Kwak, S. Impaired nucleoporins are present in sporadic amyotrophic lateral sclerosis motor neurons that exhibit mislocalization of the 43-kDa TAR DNA-binding protein. J. Clin. Neurol. 2019, 15, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Ito, H.; Hirano, A.; Fujita, K.; Wate, R.; Nakamura, M.; Kaneko, S.; Nakano, S.; Kusaka, H. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 1184–1192. [Google Scholar] [CrossRef]
- Shang, J.; Yamashita, T.; Nakano, Y.; Morihara, R.; Li, X.; Feng, T.; Huang, Y.; Fukui, Y.; Hishikawa, N.; Abe, K.; et al. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience 2017, 350, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef]
- Solomon, D.A.; Stepto, A.; Au, W.H.; Adachi, Y.; Diaper, D.C.; Hall, R.; Rekhi, A.; Boudi, A.; Tziortzouda, P.; Lee, Y.-B.; et al. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-α mediates C9orf72-related neurodegeneration. Brain 2018, 141, 2908–2924. [Google Scholar] [CrossRef]
- Coyne, A.N.; Zaepfel, B.L.; Hayes, L.; Fitchman, B.; Salzberg, Y.; Luo, E.-C.; Bowen, K.; Trost, H.; Aigner, S.; Rigo, F.; et al. G4C2 Repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron 2020, 107, 1124–1140. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Rossoll, W. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Coyne, A.N.; Baskerville, V.; Zaepfel, B.L.; Dickson, D.W.; Rigo, F.; Bennett, F.; Lusk, C.P.; Rothstein, J.D. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci. Transl. Med. 2021, 13, eabe1923. [Google Scholar] [CrossRef]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., 3rd; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Özdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.; Cook, C.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Daughrity, L.M.; Castanedes-Casey, M.; Kurti, A.; Stankowski, J.N.; Disney, M.D.; et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener. 2019, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Rabut, G.; Doye, V.; Ellenberg, J. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol. 2004, 6, 1114–1121. [Google Scholar] [CrossRef]
- Coyne, A.N.; Rothstein, J.D. Nuclear lamina invaginations are not a pathological feature of C9orf72 ALS/FTD. Acta Neuropathol. Commun. 2021, 9, 45. [Google Scholar] [CrossRef]
- Vatsavayai, S.C.; Nana, A.L.; Yokoyama, J.S.; Seeley, W.W. C9orf72-FTD/ALS pathogenesis: Evidence from human neuropathological studies. Acta Neuropathol. 2019, 137, 1–26. [Google Scholar] [CrossRef]
- Lin, Y.C.; Kumar, M.S.; Ramesh, N.; Anderson, E.N.; Nguyen, A.T.; Kim, B.; Cheung, S.; McDonough, J.A.; Skarnes, W.C.; Lopez-Gonzalez, R.; et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat. Neurosci. 2021, 24, 1077–1088. [Google Scholar] [CrossRef]
- Khosravi, B.; Hartmann, H.; May, S.; Möhl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [CrossRef]
- Steyaert, J.; Scheveneels, W.; Vanneste, J.; Van Damme, P.; Robberecht, W.; Callaerts, P.; Bogaert, E.; Van Den Bosch, L. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum. Mol. Genet. 2018, 27, 4103–4116. [Google Scholar] [CrossRef]
- Archbold, H.C.; Jackson, K.L.; Arora, A.; Weskamp, K.; Tank, E.M.H.; Li, X.; Miguez, R.; Dayton, R.D.; Tamir, S.; Klein, R.L.; et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 2018, 8, 4606. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kasper, L.H.; Mantcheva, R.T.; Mantchev, G.T.; Springett, M.J.; van Deursen, J.M. Disruption of the FG nucleoporin NUP98 causes selective changes in nuclear pore complex stoichiometry and function. Proc. Natl. Acad. Sci. USA 2001, 98, 3191–3196. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Alves, A.; Pickersgill, H.; Loïodice, I.; Hetzer, M.; Galy, V.; Hülsmann, B.B.; Köcher, T.; Wilm, M.; Allen, T.; et al. The conserved Nup107-160 complex is critical for nuclear pore complex assembly. Cell 2003, 113, 195–206. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Semmelink, M.F.W.; Steen, A.; Veenhoff, L.M. Measuring and interpreting nuclear transport in neurodegenerative disease—The example of C9orf72 ALS. Int. J. Mol. Sci. 2021, 22, 9217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 2018, 173, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Vercruysse, T.; Boeynaems, S.; Sicart, A.; Van Damme, P.; Daelemans, D.; Van Den Bosch, L. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci. Rep. 2019, 9, 15728. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Vercruysse, T.; Van Damme, P.; Van Den Bosch, L.; Daelemans, D. Quantitative nucleocytoplasmic transport assays in cellular models of neurodegeneration. Bio-Protocol 2020, 10, e3659. [Google Scholar] [CrossRef] [PubMed]
- Mahboubi, H.; Seganathy, E.; Kong, D.; Stochaj, U. Identification of novel stress granule components that are involved in nuclear transport. PLoS ONE 2013, 8, e68356. [Google Scholar] [CrossRef]
- Fujimura, K.; Suzuki, T.; Yasuda, Y.; Murata, M.; Katahira, J.; Yoneda, Y. Identification of importin α1 as a novel constituent of RNA stress granules. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2010, 1803, 865–871. [Google Scholar] [CrossRef]
- Chang, W.-L.; Tarn, W.-Y. A role for transportin in deposition of TTP to cytoplasmic RNA granules and mRNA decay. Nucleic Acids Res. 2009, 37, 6600–6612. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J.; Miki, T.; Takano, K.; Maruhashi, M.; Uchikawa, M.; Tachibana, T.; Yoneda, Y. Nuclear RNA export factor 7 is localized in processing bodies and neuronal RNA granules through interactions with shuttling hnRNPs. Nucleic Acids Res. 2008, 36, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Vercruysse, T.; Boeynaems, S.; Van Damme, P.; Daelemans, D.; Van Den Bosch, L. Stress induces nucleocytoplasmic transport deficits independent of stress granules. 2021. submitted. [Google Scholar]
- Biogen. A Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of BIIB100. 2019. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03945279?term=biib100&rank=1 (accessed on 18 December 2020).
- Guo, L.; Fare, C.M.; Shorter, J. Therapeutic dissolution of aberrant phases by nuclear-import receptors. Trends Cell Biol. 2019, 29, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Güttinger, S.; Laurell, E.; Kutay, U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 2009, 10, 178–191. [Google Scholar] [CrossRef]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R., III; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Pujol, G.; Söderqvist, H.; Radu, A. Age-associated reduction of nuclear protein import in human fibroblasts. Biochem. Biophys. Res. Commun. 2002, 294, 354–358. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Proteins | Observations | Models | Refs |
|---|---|---|---|
| Importins | |||
| Importin-β1 | Irregular, disrupted at nuclear membrane, increased cytoplasmic localization | sALS, spinal cord | [84] |
| Nuclear depletion | sALS, spinal cord | [85] | |
| No abnormalities | mutant C9orf72 ALS | [87] | |
| Importin-α1 | Nuclear depletion and cytoplasmic accumulation | mutant C9orf72 and sFTD | [88] |
| Importin-α4 | Nuclear depletion and cytoplasmic accumulation | mutant C9orf72 and sFTD | [88] |
| Nups—TM | |||
| Gp210 | Abnormal nuclear precipitations and cytoplasmic upregulation | sALS, spinal cord | [86] |
| Pom121 | Reduced expression (together with nup50, tpr, nup98, ndr1, nup107, nup133) | mutant C9orf72, motor cortex and spinal cord | [89] |
| Nups—scaffolds | |||
| Nup107 | Aggregates at nuclear membrane | mutant C9orf72 ALS, motor cortex | [83] |
| Nup205 | Abnormal nuclear localization | mutant C9orf72 ALS, motor cortex | [83] |
| Loss of immunoreactivity and large cytoplasmic inclusions | mutant TDP-43 + sALS, motor cortex | [90] | |
| Abnormal perinuclear punctate staining | mutant C9orf72, motor cortex | [90] | |
| No abnormalities | mutant SOD1, motor cortex | [90] | |
| Nups—linkers | |||
| Nup88 | Tortuous and redundant nuclear contours | mutant SOD1 ALS + sALS, spinal cord | [85] |
| Nups—central channel | |||
| Nup62 | Irregular disrupted nuclear membrane, aggregates, nuclear localization | sALS, spinal cord | [84] |
| Tortuous and redundant nuclear contours | mutant SOD1 + sALS, spinal cord | [85] | |
| Nup—outer structures | |||
| Nup50 | Abnormal nuclear precipitations and cytoplasmic upregulation | sALS, spinal cord | [86] |
| Ran gradient | |||
| RanGap | Abnormal nuclear localization | mutant C9orf72 ALS, motor cortex | [83] |
| Abnormal nuclear precipitations and cytoplasmic upregulation | SALS, spinal cord | [86] | |
| No abnormalities | mutant TDP-43 + mutant C9orf72 + SALS motor cortex | [90] | |
| No abnormalities | mutant C9orf72 ALS | [87] | |
| In Vitro | |||
| Proteins | Observations | Models | Refs |
| Importins | |||
| TPNO3 | No abnormal staining | Patient-derived mutant C9orf72 induced neurons | [92] |
| Importin-α3 | No abnormal staining | Patient-derived mutant C9orf72 induced neurons | [92] |
| Exportins | |||
| Exportin 5 | No abnormal staining | Patient-derived mutant C9orf72 induced neurons | [92] |
| Nups | |||
| Mab414 (nup62, nup153›, nup214, nup358) | Abnormal staining | Mutant TDP-43 iPSC motor neurons | [90] |
| NPC proteins | Interaction with PR50 and GR50 | HEK293T cells expressing PR50 or GR50 | [41] |
| Gle1, nup88, nup214, nup358, nup35, nup93, nup58, nup62, nup98, nup107, nup155, nup160 nup205, nup153, aladin, nxf1 | Aggregation together with TDP-43 CTF | BioID approach in Neuro-2a cells expressing TDP-43 CTF | [90] |
| Nups—TM | |||
| Pom121 | Mislocalization upon TDP-CTF OE | Neuro-2a cells cotransfected with plasmid expressing pom121 and TDP-43 CTF | [90] |
| Reduced expression | Patient-derived mutant C9orf72 iPSC induced neurons | [89] | |
| Gp210 | Mislocalization upon TDP-CTF OE | Neuro-2a cells cotransfected with plasmid expressing gp210 and TDP-43 CTF | [90] |
| Nups—scaffolds | |||
| Nup205 | Abnormal staining | Mutant TDP-43 and mutant C9orf72 patient fibroblasts | [90] |
| No abnormal staining | Sporadic ALS patient fibroblasts | [90] | |
| Nups—central channel | |||
| Nup62 | Irregular nuclear contour | Spinal cord of SOD1G93A mouse model | [85] |
| Nup98 | Disturbed distribution | Neuro-2a cells transfected with TDP-43 CTF | [90] |
| Lamina | |||
| Lamina B1 | Abnormal staining | Mutant TDP-43 and mutant C9orf72 patient fibroblasts | [90] |
| No abnormal staining | Sporadic patient fibroblasts | [90] | |
| Abnormal staining | Mutant TDP-43 iPSC motor neurons | [90] | |
| Abnormal staining | Mouse primary cortical neurons expressing TDP-43 CTF/TDP-43Q331K/TDP-43M337V | [90] | |
| No abnormal staining | Patient-derived mutant C9orf72 induced neurons | [92] | |
| Ran gradient | |||
| RanGAP | Nuclear puncta | S2 Drosophila cells transfected with (G4C2)30 | [83] |
| Nuclear puncta | Mutant C9orf72 iPSC-derived neurons | [83] | |
| Abnormal staining | Mouse primary cortical neurons expressing TDP 43 CTF or TDP-43-mtNLS | [90] | |
| No abnormal staining | Mouse primary cortical neurons expressing TDP 43Q331K or TDP 43M337V | [90] | |
| No abnormal staining | Patient-derived mutant C9orf72 induced neurons | [92] | |
| RanGEF | Nuclear depletion | Patient derived mutant C9orf72 induced neurons | [92] |
| Ran | Reduced nuclear/cytoplasmic ratio | S2 Drosophila cells transfected with (G4C2)30 | [83] |
| Reduced nuclear/cytoplasmic ratio | Patient-derived mutant C9orf72 induced neurons | [83] | |
| No change in nuclear/cytopIasmic ratio | Mouse primary cortical neurons expressing TDP-43 CTF or TDP-43-mtNLS | [90] | |
| RAN export | |||
| Thoc2 | Cytoplasmic mislocalization | HEK293T cells expressing TDP-43 fragments | [93] |
| In vivo | |||
| Proteins | Observations | Models | Refs |
| Importins | |||
| TPNO1 | Cytoplasmic aggregates | Prp-TDP-43A315T-GFP mouse model | [94] |
| Importin-α1 | Cytoplasmic accumulation and nuclear depletion | Salivary gland cells of Drosophila expressing poly-GA64 or poly-GR64 | [88] |
| Cytoplasmic accumulation and nuclear depletion | Salivary gland cells of Drosophila expressing ΔNLS-TDP-43, human TDP-43 or TDP-43Q331K | [88] | |
| Cytoplasmic accumulation and nuclear depletion | Salivary gland cells of Drosophila expressing poly-GA64 or poly-GR64 | [88] | |
| Importin-α4 | Cytoplasmic accumulation and nuclear depletion | Salivary gland cells of Drosophila expressing ΔNLS-TDP-43, human TDP-43 or TDP-43Q331K | [88] |
| Nups | |||
| Mab414 | No abnormal staining | Salivary gland cells of Drosophila expressing poly-GA64 or poly-GR64 | [88] |
| (nup62, nup153, nup214, nup358) | No abnormal staining | Salivary gland cells of Drosophila expressing ΔNLS-TDP-43, human TDP-43 or TDP-43Q331K | [88] |
| Nups—TM | |||
| Gp210 | Increased nuclear localization | Spinal cord motor neurons of SOD1G93A mouse model | [86] |
| Nups—Scaffolds | |||
| Nup107 | Inclusions near nuclear envelope | Salivary gland cells of Drosophila expressing (G4C)58 | [35] |
| Increased nuclear localization | Spinal cord motor neurons of SOD1G93A mouse model | [86] | |
| Nup205 | Increased nuclear localization | Spinal cord motor neurons of SOD1G93A mouse model | [86] |
| Nups—outer structures | |||
| Nup50 | No abnormal staining | Salivary gland cells of Drosophila expressing poly-GA 64 or poly-GR64 | [88] |
| Lamina | |||
| Lamina C | Abnormal nuclear membrane | Salivary gland cells of Drosophila expressing (G4C2)58 | [35] |
| Ran gradient | |||
| RanGap | Increased nuclear localization | Spinal cord motor neurons of SOD1G93A mouse model | [86] |
| No abnormal staining | Salivary gland cells of Drosophila expressing poly-GA64 or poly-GR64 | [88] | |
| Increased nuclear localization | PrP-TDP43A315T GFP mouse model | [94] | |
| Increased localization to nuclear invaginations | Mice expressing (G4C2)149 via intracerebroventricular injections with AAV | [95] | |
| Proteins | E/S | Observations | Models | Refs |
|---|---|---|---|---|
| Importins | ||||
| Importin-β1 | E | LOF enhances phenotype | Drosophila expressing poly-GR50 | [41] |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| Importin-α1 | S | OE enhances phenotype | Drosophila (G4C2)30—no DPRs detected | [83] |
| S | LOF suppresses phenotype | Drosophila expressing poly-GR50 | [41] | |
| Importin-α3 | E | OE suppresses phenotype | Yeast expressing poly-PR50 | [92] |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| E | OE suppresses NC-transport phenotype | HeLa cells expressing GA149 | [100] | |
| Importin-α4 | E | LOF enhances phenotype | Drosophila expressing poly-GR50 | [41] |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| E | OE suppresses NC-transport phenotype | HeLa cells expressing GA149 | [100] | |
| TPNO1 | E | LOF enhances phenotype | Drosophila (G4C2)58—DPRs detected | [35] |
| E | OE suppresses phenotype | Yeast expressing poly-PR50 | [92] | |
| E | LOF enhances phenotype | Drosophila expressing poly-GR50 | [41] | |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| TPNO3 | E | OE suppresses phenotype | Yeast expressing poly-PR50 | [92] |
| Importin 4 | S | LOF suppresses phenotype | Drosophila expressing poly-PR25 | [37] |
| Importin 5 | S | LOF suppresses phenotype | Drosophila expressing poly-GR50 | [41] |
| Importin 7 | S | LOF suppresses phenotype | Drosophila expressing poly-GR50 | [41] |
| Importin 9 | E | OE suppresses phenotype | Yeast expressing poly-PR50 | [92] |
| Importin 11 | E | OE suppresses phenotype | Yeast expressing poly-PR50 | [92] |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| Exportins | ||||
| Exportin 1 | S | LOF suppresses phenotype–confirmed by KPT-276 | Drosophila (G4C2)30—no DPRs detected | [83] |
| S | LOF suppresses phenotype | Drosophila expressing human FUSR521G | [101] | |
| E | LOF enhances phenotype—confirmed by LMB | Drosophila (G4C2)58 - DPRs detected | [35] | |
| E | LOF enhances phenotype | Drosophila expressing poly-GR50 | [41] | |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| S | 2.5 nM KPT-350/KPT-335 is mildly protective (no impaired transport) | Rat cortical neurons OE human TDP-43WT | [102] | |
| E | 10 nM KPT-350/KPT-335 is toxic | Rat cortical neurons OE human TDP-43WT | [102] | |
| KPT-350 induces a partial rescue limited by weight loss | Rat model AAV9 brain injected TDP-43 mRNA | [102] | ||
| S | KPT-335 treatment suppresses phenotype | Mouse cortical neurons expressing TDP-43 CTF or TDP-43Q331K | [90] | |
| S | 1 µM KPT-335/KPT-276 treatment suppresses phenotype | Drosophila TDP-43WT or TDP-43Q331K OE | [90] | |
| E | 150 nM KPT335/KPT-276 treatment enhances phenotype | Mouse cortical neurons expressing TDP-43 CTF or TDP-43Q331K | [90] | |
| E | 5 µM KPT-335/KPT-276 | Drosophila TDP-43WT or TDP-43Q331K OE | [90] | |
| Exportin 5 | E | OE suppresses phenotype | Yeast expressing poly-PR50 | [92] |
| Nups—TM | ||||
| Ndc1 | S | OE enhances phenotype | Yeast expressing poly-PR50 | [92] |
| Nups—scaffolds | ||||
| Nup107 | S | LOF suppresses phenotype | Drosophila (G4C2)58 - DPRs detected | [35] |
| S | LOF suppresses phenotype | Drosophila TDP-43WT or TDP-43Q331K OE | [90] | |
| S | LOF suppresses phenotype | Drosophila expressing poly-PR25 | [37] | |
| Nup160 | S | LOF suppresses phenotype | Drosophila (G4C2)58 - DPRs detected | [35] |
| Nup205 | S | LOF suppresses phenotype | Drosophila expressing poly-GR50 | [41] |
| Seh1 | E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] |
| Nup155 | S | LOF suppresses phenotype | Drosophila expressing poly-PR25 | [37] |
| S | LOF suppresses phenotype | Drosophila expressing human FUSR521G | [101] | |
| Nups—linkers | ||||
| Nup93 | S | LOF suppresses phenotype | Drosophila TDP-43WT or TDP-43Q331K OE | [90] |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| Nups—central channel | ||||
| Nup96—Nup98 | S | LOF suppresses phenotype | Drosophila (G4C2)58 - DPRs detected | [35] |
| S | LOF suppresses phenotype | Drosophila TDP-43WT or TDP-43Q331K OE | [90] | |
| S | LOF suppresses phenotype | Drosophila FUSWT, FUSR518K, FUSR521C OE | [99] | |
| Nup62 | E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] |
| S | LOF suppresses phenotype | Drosophila FUSWT, FUSR518K, FUSR521C OE | [99] | |
| S | OE enhances phenotype | Drosophila FUSWT, FUSR518K, FUSR521C OE | [99] | |
| E | OE suppresses NC-transport phenotype | HeLa cells expressing GA149 | [100] | |
| Nup54 | E | OE suppresses NC-transport phenotype | HeLa cells expressing GA149 | [100] |
| S | LOF suppresses phenotype | Drosophila FUSWT, FUSR518K, FUSR521C OE | [99] | |
| Nups—outer structures | ||||
| Nup50 | E | LOF enhances phenotype | Drosophila (G4C2)58 - DPRs detected | [35] |
| S | LOF suppresses phenotype | Drosophila expressing poly-PR25 | [37] | |
| Nup153 | E | LOF enhances phenotype | Drosophila (G4C2)58—DPRs detected | [35] |
| Nup214 | S | LOF suppresses phenotype | Drosophila TDP-43WT or TDP-43Q331K OE | [90] |
| S | LOF suppresses phenotype | Drosophila FUSWT, FUSR518K, FUSR521C OE | [99] | |
| Tpr | E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] |
| RanGradient | ||||
| RanGap | E | GOF mutation/OE suppresses phenotype | Drosophila (G4C2)30—no DPRs detected | [83] |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| RanGEF | S | OE enhances phenotype | Drosophila (G4C2)30—no DPRs detected | [83] |
| S | OE enhances phenotype | Yeast expressing poly-PR50 | [92] | |
| E | LOF enhances phenotype | Drosophila expressing poly-PR25 | [37] | |
| Ran | E | Dominant negative mutation enhances phenotype | Drosophila (G4C2)58—DPRs detected | [35] |
| RAN export | ||||
| Alyref | S | LOF suppresses phenotype | Drosophila (G4C2)58—DPRs detected | [35] |
| Gle1 | E | LOF enhances phenotype | Drosophila (G4C2)58—DPRs detected | [35] |
| Assay | Models | Observations | Suggested Mechanism | Refs |
|---|---|---|---|---|
| Imp-β1/α1 | ||||
| NLS-NES-GFP | Drosophila salivary glands expressing (G4C2)30 repeats | Reduced N/C ratio | RanGap sequestration in nuclear RNA foci | [107] |
| Mutant C9orf72 iPSC-derived neurons | Reduced import based on FRAP | |||
| GFP-NLS-NES | U2OS treated with PR20 peptides | Reduced import over time | Pores are blocked by poly-PR peptides | [45] |
| BSA-NLS | Digitonin-treated HeLa cells exposed to PR20 peptides | Reduced import | ||
| RFP-NLSTDP43 | HeLa cells transfected with GA149 expressing plasmid | Increased cytoplasmic levels | Cytoplasmic poly-GA aggregates | [100] |
| HeLa cells transfected with GR149 expressing plasmid | No consistent differences observed | n.a. | ||
| HeLa cells transfected with PR175 expressing plasmid | No differences observed | n.a. | ||
| NES-tdTomato-NLS | Primary mouse cortical neurons expressing TDP-43 CTF/mTDP-43 | Reduced N/C ratio | TDP-43 aggregates sequester NC-transport proteins | [90] |
| Fibroblasts of mutant C9orf72, TDP-43 and sALS patients | ||||
| HEK293T cells transfected with PR50/GR50 expressing plasmids | Mislocalization of reporter | Stress granules sequester NC-transport proteins | [107] | |
| ALS-FUS human spinal neurons and isogenic controls | Decreased nuclear import | Increased interaction of mutant FUS with Nup62 | [99] | |
| Artificial importin-βcargo based on FRET | Permeabilized HeLa cells incubated with PR20 and GR20 peptides | Decreased nuclear import | Poly-PR and poly-GR bind and disrupt cargo loading of importin-β | [47] |
| Fluorescent dextrans | Permeabilized HeLa cells incubated with PR20 and GR20 peptides | Increased passive transport | ||
| Hormone-induced import assay: GCR2-GFP2-TDP43 or GCR2-GFP2-(MBP)-cNLS | HeLa cells incubated with TMR-GR25 peptides | Reduced import | Reduced solubility of importin-α/β via poly-GR binding | [46] |
| HeLa cells incubated with TMR-PR25 peptides | No difference observed | n.a. | ||
| NLSSV40-mNeonGreen2x-NESpki | HeLa cells incubated with GR20 or PR20 +/− leptomycin B (LMB) | No differences observed | n.a. | [108] |
| HeLa cells transduced with GR100 or PR100 +/− LMB | No differences observed | n.a. | ||
| SH-SY5Y cells transduced with GR100 or PR100 +/− LMB | No differences observed | n.a. | ||
| iPSC-derived MNs transduced with GR100 or PR100 +/− LMB | No differences observed | n.a. | ||
| NLSc-myc-GFP2x-NESikb2 | HeLa cells transduced with GR100 or PR100 | No differences observed | n.a. | |
| TPNO | ||||
| RFP-NLSpY(hnRNPA1) | HeLa cells transfected with GA149, GR149 or PR175 expressing plasmids | No differences observed | n.a. | [100] |
| YFP-M9-CFP | Digitonin treated HeLa cells incubated with PR20 and GR20 peptides | Decreased nuclear import | Poly-PR and poly-GR bind and disrupt cargo loading of TPNO1 | [47] |
| NESpki-mNeonGreen2x-NLSFUS | HeLa cells incubated with GR20 or PR20 | No differences observed | n.a. | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanneste, J.; Van Den Bosch, L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12175. https://doi.org/10.3390/ijms222212175
Vanneste J, Van Den Bosch L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(22):12175. https://doi.org/10.3390/ijms222212175
Chicago/Turabian StyleVanneste, Joni, and Ludo Van Den Bosch. 2021. "The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 22: 12175. https://doi.org/10.3390/ijms222212175
APA StyleVanneste, J., & Van Den Bosch, L. (2021). The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 22(22), 12175. https://doi.org/10.3390/ijms222212175