
Nicotinamide Mononucleotide Prevents Free Fatty Acid-Induced Reduction in Glucose Tolerance by Decreasing Insulin Clearance

 , , , ,
, , , , 

Abstract
1. Introduction
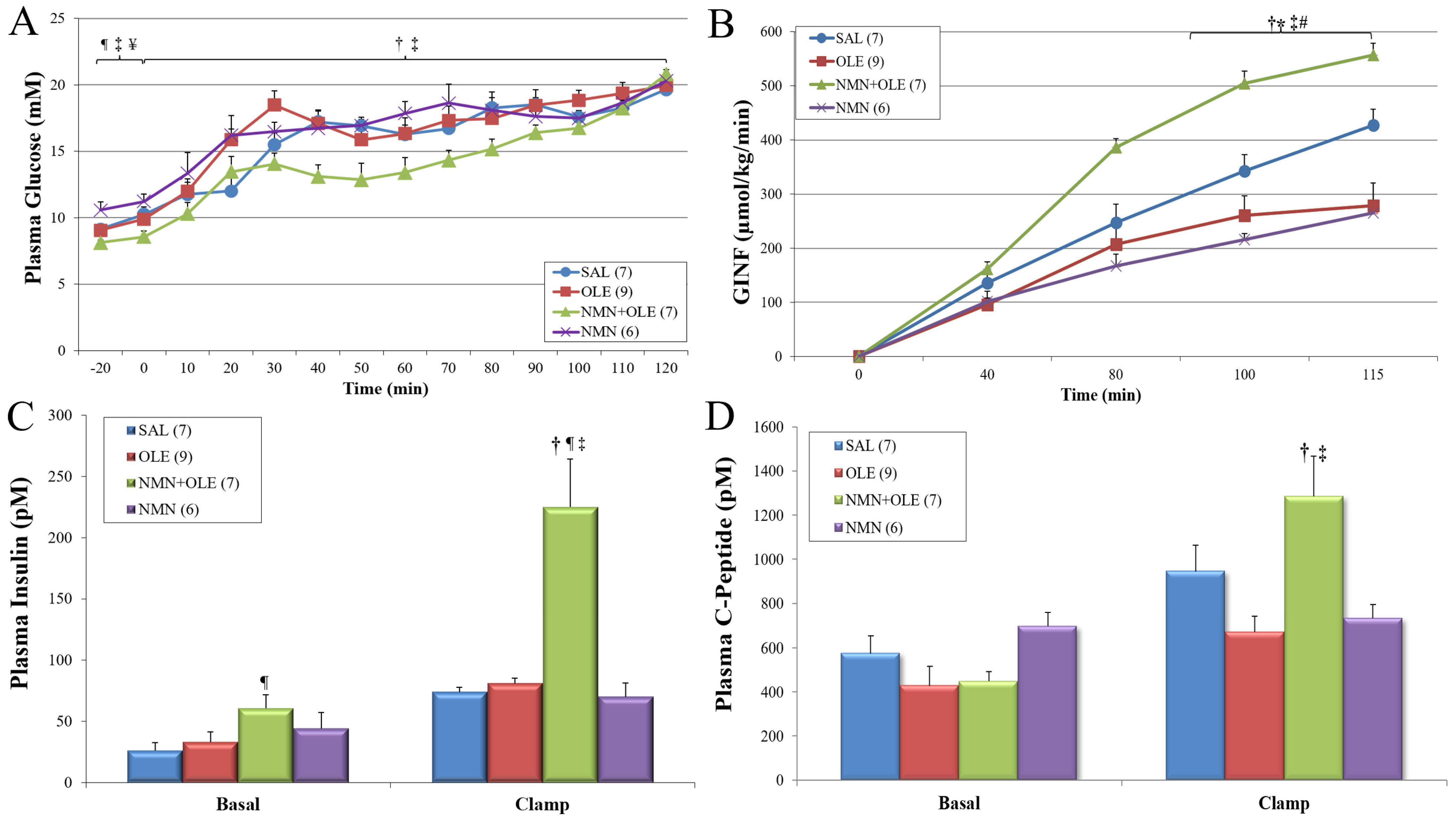
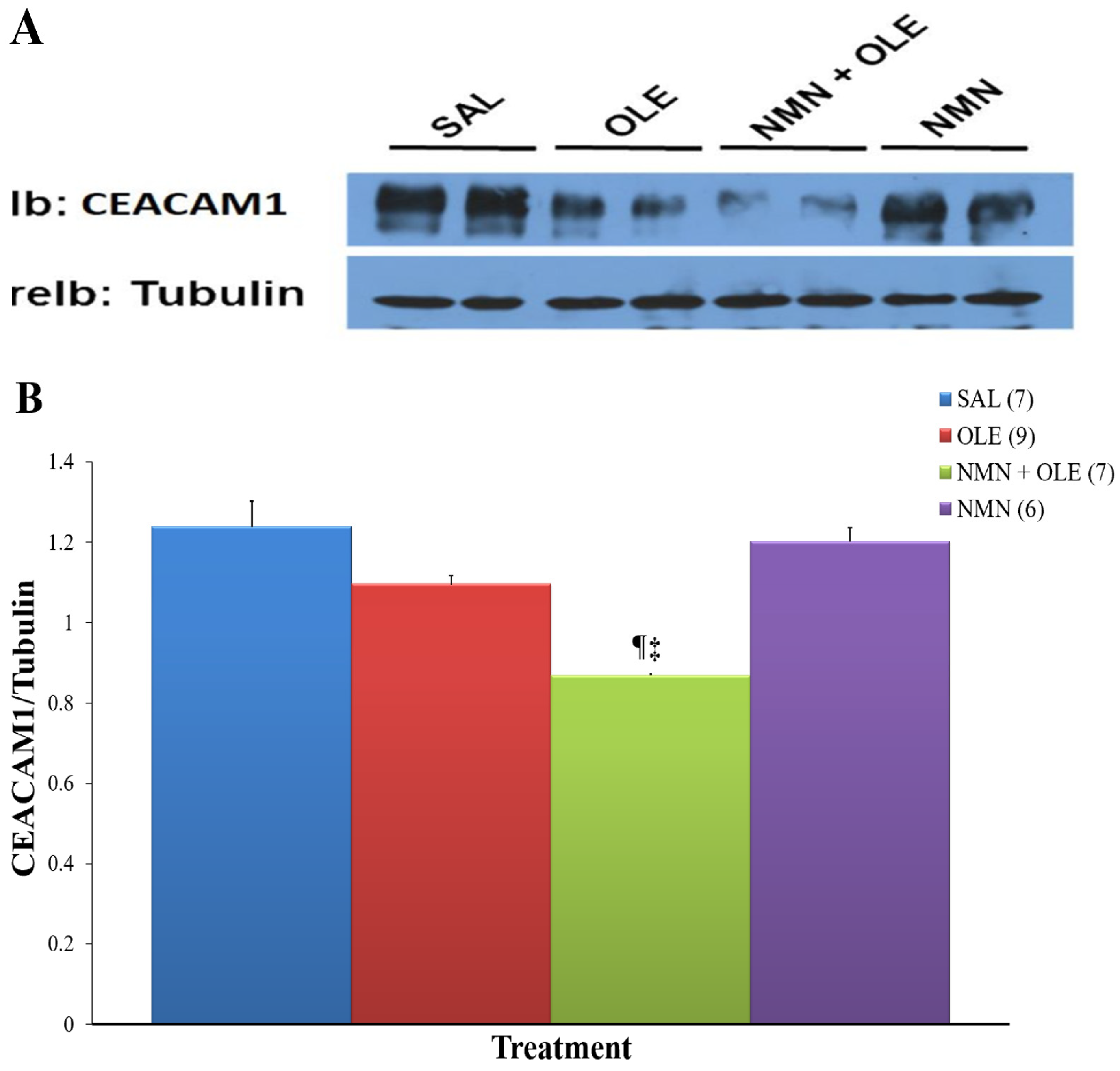
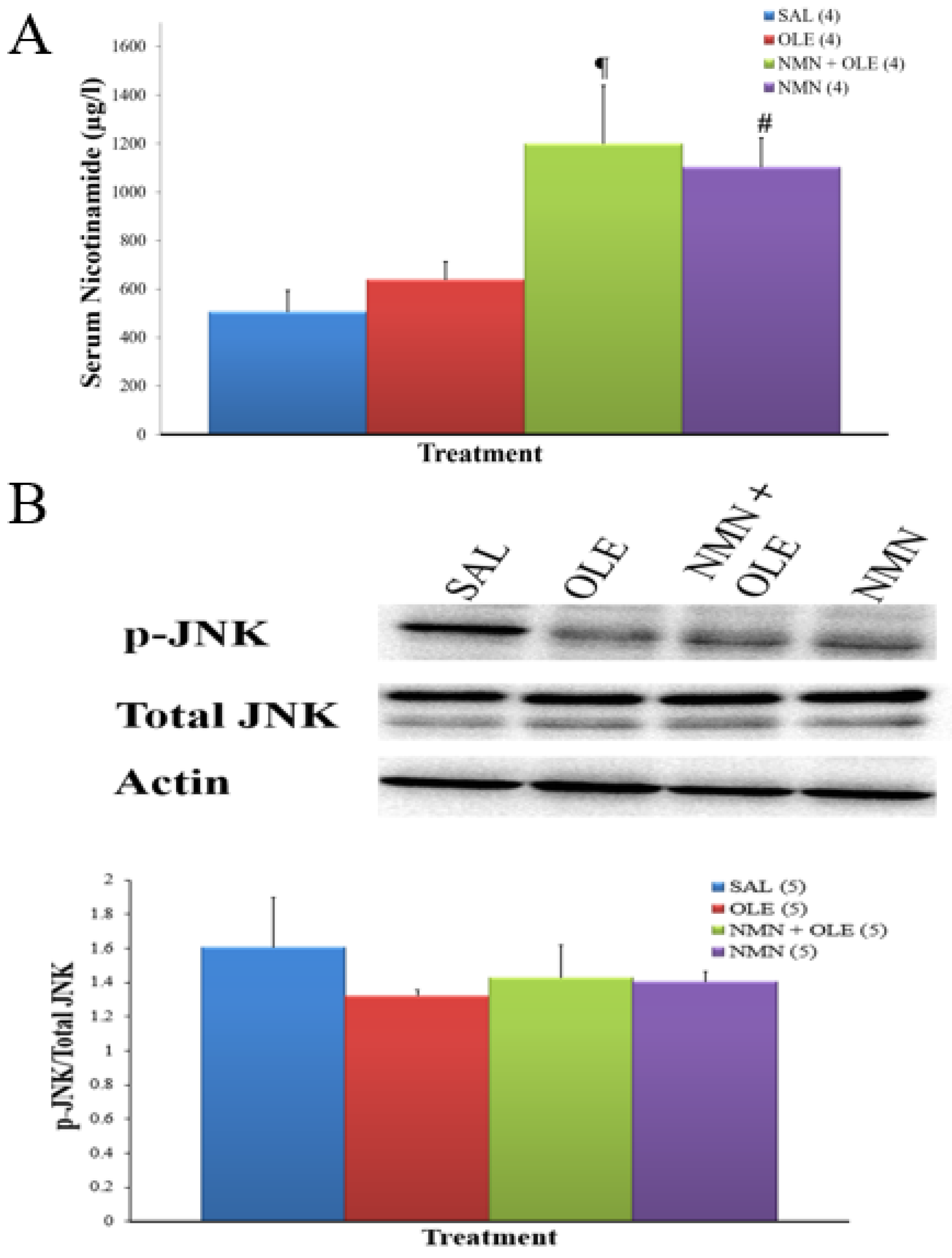
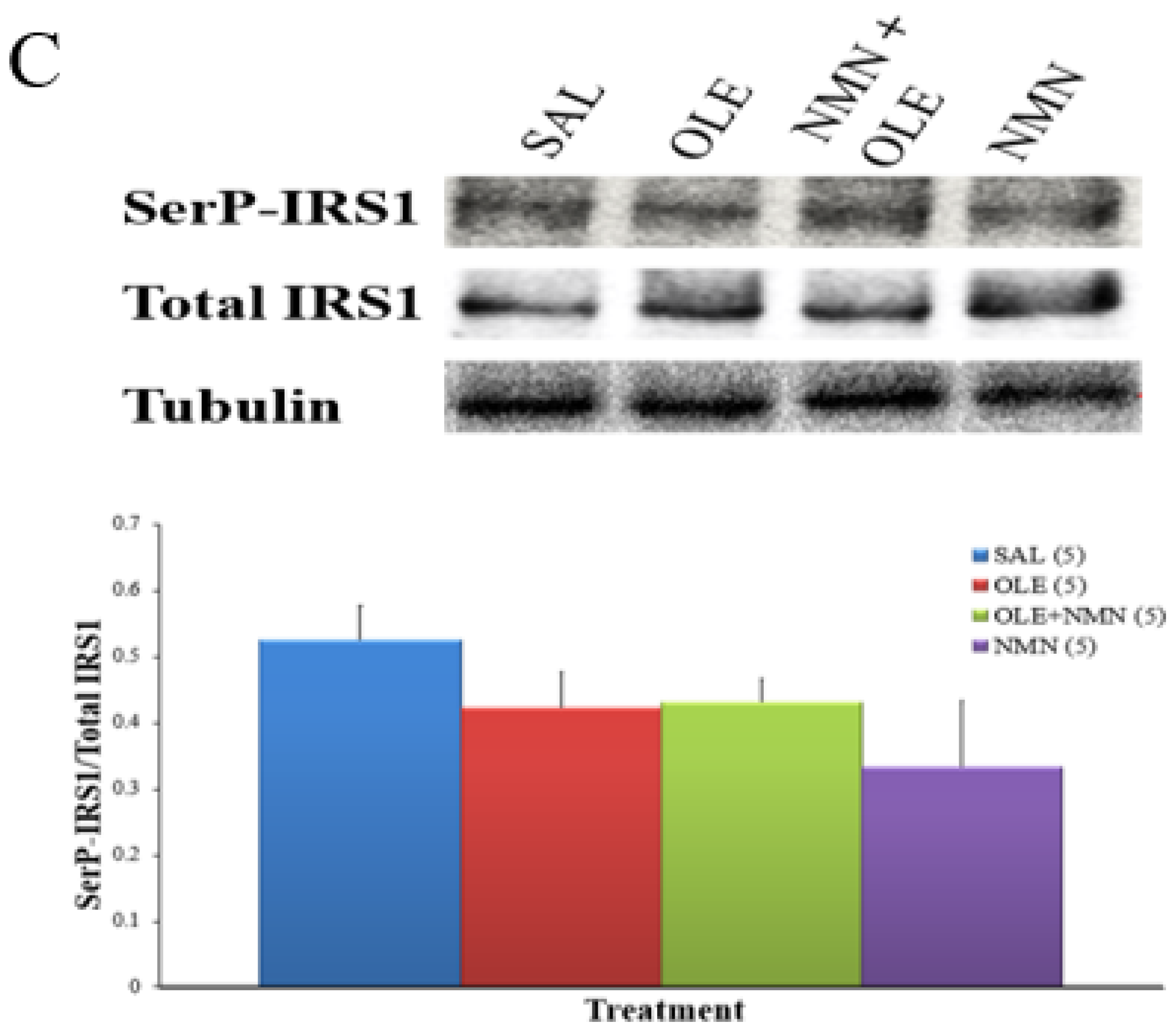
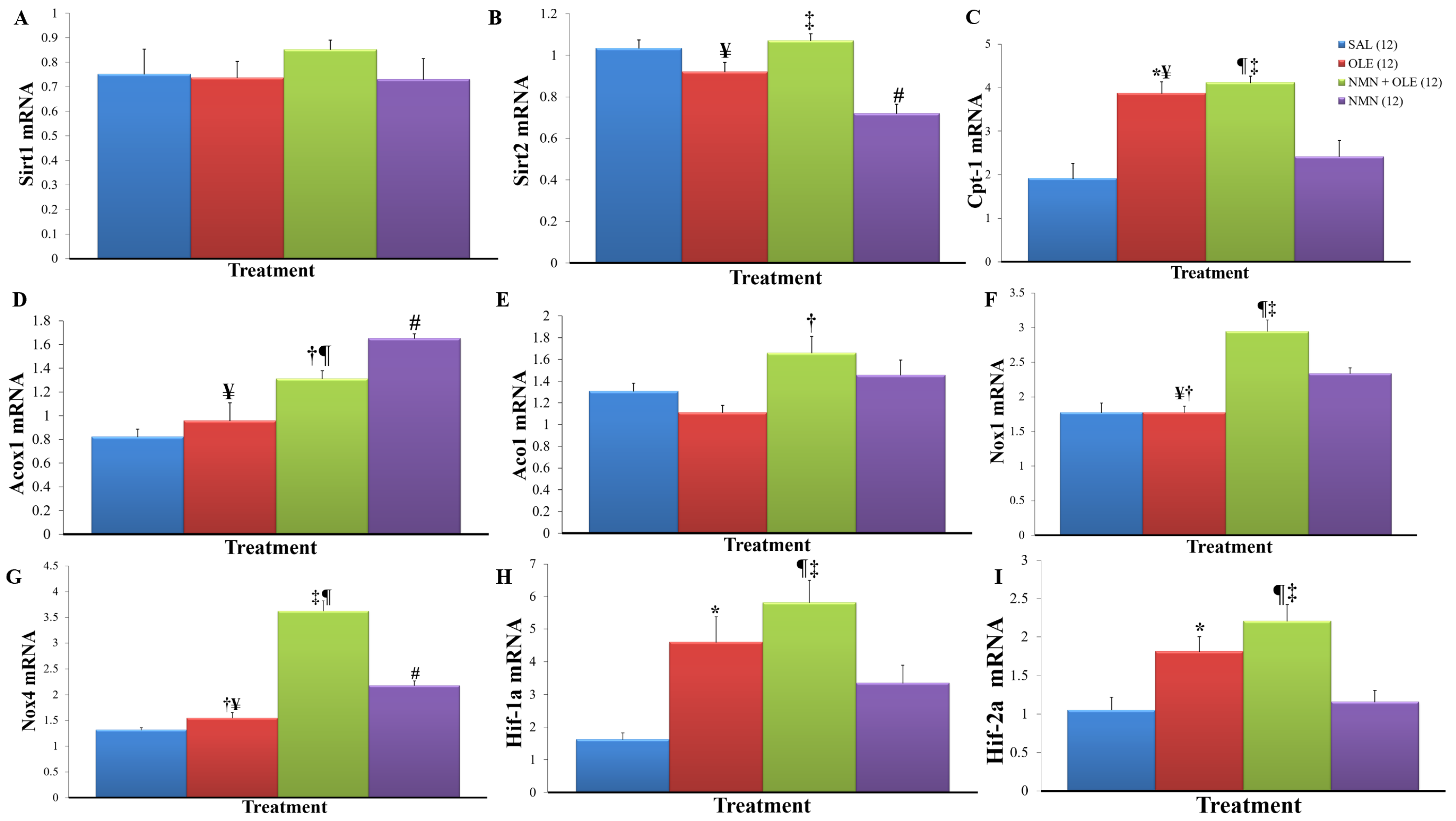
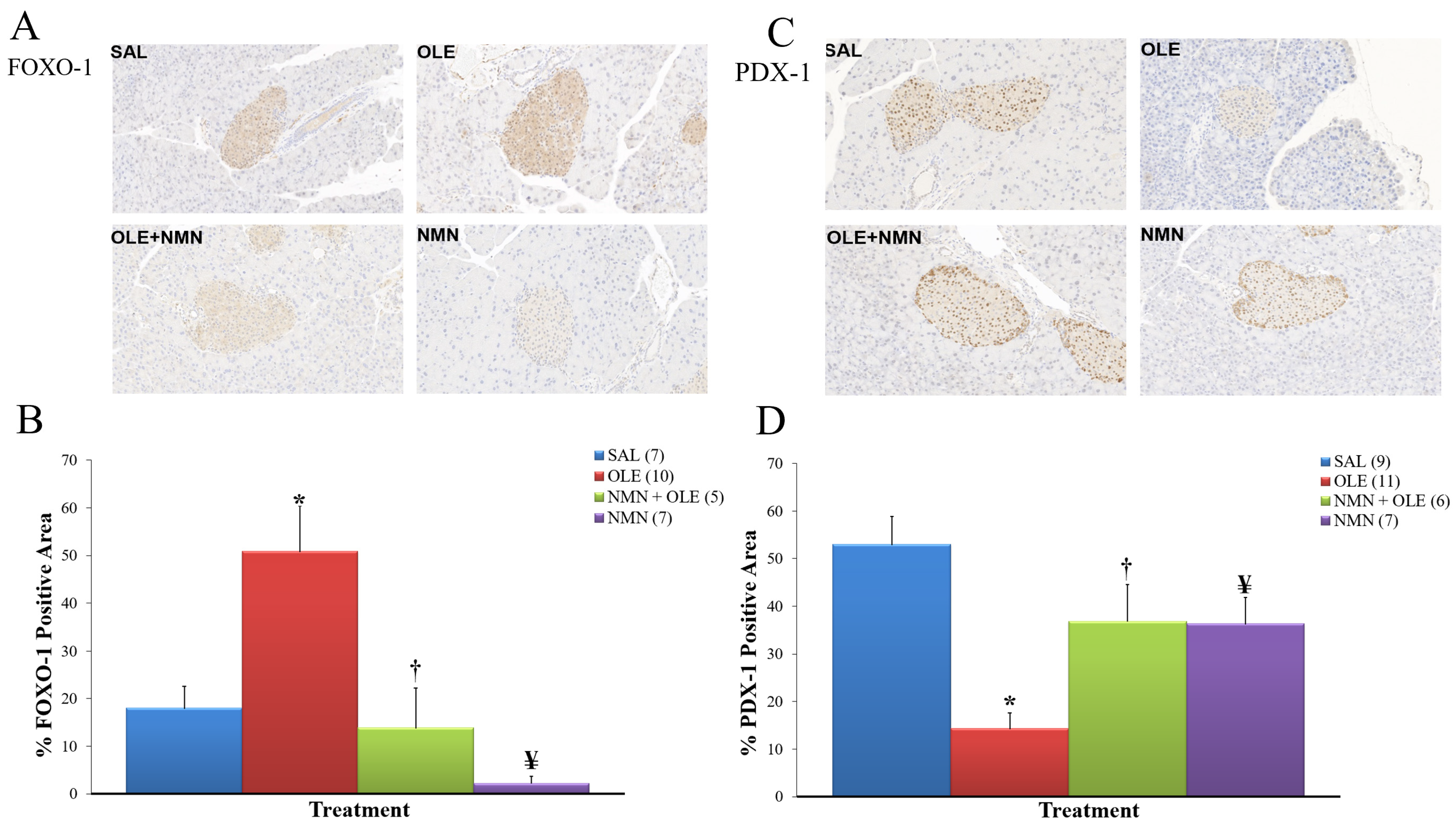
2. Results
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Mouse Cannulation Surgery
4.3. Intravenous Infusion of Mice with NMN and Oleate
4.4. Hyperglycemic Clamp
4.5. Plasma Assays
4.6. Immunohistochemistry
4.7. Western Blot Analysis
4.8. Hepatic NAD+ and NADH Assay
4.9. Studies in HepG2 Cells
4.10. Semi-Quantitatve RT-PCR
4.11. Calculations
4.11.1. Insulin Sensitivity Index
4.11.2. Disposition Index
4.11.3. Insulin Clearance Index
4.11.4. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boden, G. Obesity, Insulin Resistance and Free Fatty Acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef]
- Sato, H.; Terasaki, T.; Mizuguchi, H.; Okumura, K.; Tsuji, A. Receptor-Recycling Model of Clearance and Distribution of Insulin in the Perfused Mouse Liver. Diabetologia 1991, 34, 613–621. [Google Scholar] [CrossRef]
- Rabkin, R.; Ryan, M.P.; Duckworth, W.C. The Renal Metabolism of Insulin. Diabetologia 1984, 27, 351–357. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient Control of Glucose Homeostasis through a Complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.-W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in Cellular Functions: Role of Polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef]
- Desai, T.; Koulajian, K.; Ivovic, A.; Breen, D.M.; Luu, L.; Tsiani, E.L.; Wheeler, M.B.; Giacca, A. Pharmacologic or Genetic Activation of SIRT1 Attenuates the Fat-Induced Decrease in Beta-Cell Function In Vivo. Nutr. Diabetes 2019, 9, 11. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Klein, S. Effect of “Nicotinamide Mononucleotide” (NMN) on Cardiometabolic Function (NMN); Washington University School of Medicine: St. Louis, MO, USA, 2017. [Google Scholar]
- Itoh, H. Assessment of the Safety of Nicotinamide Mononucleotide (NMN) in Healthy Subjects; Phase I Study; Keio University School of Medicine: Tokyo, Japan, 2016. [Google Scholar]
- Itoh, H. Assessment of the Safety of Long-Term Nicotinamide Mononucleotide (NMN) in Healthy Subjects; Phase II Study. In The Clinical Trial to Evaluate Metabolic-Syndrome-Related Parameters to Develop NMN as Foods with Function Claims; Keio University School of Medicine: Tokyo, Japan, 2017. [Google Scholar]
- Pereira, S.; Park, E.; Moore, J.; Faubert, B.; Breen, D.M.; Oprescu, A.I.; Nahle, A.; Kwan, D.; Giacca, A.; Tsiani, E. Resveratrol Prevents Insulin Resistance Caused by Short-Term Elevation of Free Fatty Acids In Vivo. Appl. Physiol. Nutr. Metab. 2015, 40, 1129–1136. [Google Scholar] [CrossRef]
- Ivovic, A.; Oprescu, A.I.; Koulajian, K.; Mori, Y.; Eversley, J.A.; Zhang, L.; Nino-Fong, R.; Lewis, G.F.; Donath, M.Y.; Karin, M.; et al. IKKβ Inhibition Prevents Fat-Induced Beta Cell Dysfunction In Vitro and In Vivo in Rodents. Diabetologia 2017, 60, 2021–2032. [Google Scholar] [CrossRef]
- Koulajian, K.; Ivovic, A.; Ye, K.; Desai, T.; Shah, A.; George Fantus, I.; Ran, Q.; Giacca, A. Overexpression of Glutathione Peroxidase 4 Prevents β-Cell Dysfunction Induced by Prolonged Elevation of Lipids In Vivo. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E254–E262. [Google Scholar] [CrossRef]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. FFA-Induced Hepatic Insulin Resistance In Vivo Is Mediated by PKC, NADPH Oxidase, and Oxidative Stress. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E34–E46. [Google Scholar] [CrossRef]
- Poy, M.N.; Yang, Y.; Rezaei, K.; Fernström, M.A.; Lee, A.D.; Kido, Y.; Erickson, S.K.; Najjar, S.M. CEACAM1 Regulates Insulin Clearance in Liver. Nat. Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 Regulates Adipocyte Differentiation through FoxO1 Acetylation/Deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef]
- Buteau, J.; Shlien, A.; Foisy, S.; Accili, D. Metabolic Diapause in Pancreatic β-Cells Expressing a Gain-of-Function Mutant of the Forkhead Protein Foxo1. J. Biol. Chem. 2007, 282, 287–293. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Khuder, S.S.; Al-Share, Q.Y.; Russo, L.; Abdallah, S.L.; Patel, P.R.; Heinrich, G.; Muturi, H.T.; Mopidevi, B.R.; Oyarce, A.M.; et al. PPARα (Peroxisome Proliferator-Activated Receptor α) Activation Reduces Hepatic CEACAM1 Protein Expression to Regulate Fatty Acid Oxidation during Fasting-Refeeding Transition. J. Biol. Chem. 2016, 291, 8121–8129. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and Multiple Roles in Disease and Physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef]
- Sarikhani, M.; Mishra, S.; Desingu, P.A.; Kotyada, C.; Wolfgeher, D.; Gupta, M.P.; Singh, M.; Sundaresan, N.R. SIRT2 Regulates Oxidative Stress-Induced Cell Death through Deacetylation of c-Jun NH2-Terminal Kinase. Cell Death Differ. 2018, 25, 1638–1656. [Google Scholar] [CrossRef]
- Najjar, S.M.; Boisclair, Y.R.; Nabih, Z.T.; Philippe, N.; Imai, Y.; Suzuki, Y.; Suh, D.S.; Ooi, G.T. Cloning and Characterization of a Functional Promoter of the Rat Pp120 Gene, Encoding a Substrate of the Insulin Receptor Tyrosine Kinase. J. Biol. Chem. 1996, 271, 8809–8817. [Google Scholar] [CrossRef]
- Pereira, S.; Yu, W.Q.; Frigolet, M.E.; Beaudry, J.L.; Shpilberg, Y.; Park, E.; Dirlea, C.; Nyomba, B.L.G.; Riddell, M.C.; Fantus, I.G.; et al. Duration of Rise in Free Fatty Acids Determines Salicylate’s Effect on Hepatic Insulin Sensitivity. J. Endocrinol. 2013, 217, 31–43. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and Maladaptive Cardiorespiratory Responses to Continuous and Intermittent Hypoxia Mediated by Hypoxia-Inducible Factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-Inducible Transcription Factor 2α Promotes Steatohepatitis through Augmenting Lipid Accumulation, Inflammation, and Fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef]
- Najjar, S.M.; Yang, Y.; Fernström, M.A.; Lee, S.-J.; DeAngelis, A.M.; Rjaily, G.A.A.; Al-Share, Q.Y.; Dai, T.; Miller, T.A.; Ratnam, S.; et al. Insulin Acutely Decreases Hepatic Fatty Acid Synthase Activity. Cell Metab. 2005, 2, 43–53. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia Drives Hepatic Insulin Resistance in Male Mice with Liver-Specific Ceacam1 Deletion Independently of Lipolysis. Metabolism 2019, 93, 33–43. [Google Scholar] [CrossRef]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of Insulin Signaling Pathway in Rat Liver: mTORC1 Required for Stimulation of Lipogenesis, but Not Inhibition of Gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef]
- Kotronen, A.; Vehkavaara, S.; Seppala-Lindroos, A.; Bergholm, R.; Yki-Jarvinen, H. Effect of Liver Fat on Insulin Clearance. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1709–E1715. [Google Scholar] [CrossRef]
- Wiesenthal, S.R.; Sandhu, H.; McCall, R.H.; Tchipashvili, V.; Yoshii, H.; Polonsky, K.; Shi, Z.Q.; Lewis, G.F.; Mari, A.; Giacca, A. Free Fatty Acids Impair Hepatic Insulin Extraction In Vivo. Diabetes 1999, 48, 766–774. [Google Scholar] [CrossRef]
- Yoshii, H.; Lam, T.K.T.; Gupta, N.; Goh, T.; Haber, C.A.; Uchino, H.; Kim, T.T.Y.; Chong, V.Z.; Shah, K.; Fantus, I.G.; et al. Effects of Portal Free Fatty Acid Elevation on Insulin Clearance and Hepatic Glucose Flux. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1089–E1097. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic Drugs, Polyunsaturated Fatty Acids, and Eicosanoids Are Ligands for Peroxisome Proliferator-Activated Receptors α and δ. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Russo, L.; Ghanem, S.S.; Patel, P.R.; Oyarce, A.M.; Heinrich, G.; Najjar, S.M. Fenofibrate Decreases Insulin Clearance and Insulin Secretion to Maintain Insulin Sensitivity. J. Biol. Chem. 2016, 291, 23915–23924. [Google Scholar] [CrossRef]
- Tan, M.; Tang, C.; Zhang, Y.; Cheng, Y.; Cai, L.; Chen, X.; Gao, Y.; Deng, Y.; Pan, M. SIRT1/PGC-1α Signaling Protects Hepatocytes against Mitochondrial Oxidative Stress Induced by Bile Acids. Free Radic. Res. 2015, 49, 935–945. [Google Scholar] [CrossRef]
- Erion, D.M.; Yonemitsu, S.; Nie, Y.; Nagai, Y.; Gillum, M.P.; Hsiao, J.J.; Iwasaki, T.; Stark, R.; Weismann, D.; Yu, X.X.; et al. SirT1 Knockdown in Liver Decreases Basal Hepatic Glucose Production and Increases Hepatic Insulin Responsiveness in Diabetic Rats. Proc. Natl. Acad. Sci. USA 2009, 106, 11288–11293. [Google Scholar] [CrossRef]
- Imai, S. A Possibility of Nutriceuticals as an Anti-Aging Intervention: Activation of Sirtuins by Promoting Mammalian NAD Biosynthesis. Pharmacol. Res. 2010, 62, 42–47. [Google Scholar] [CrossRef]
- Imai, T.; Anderson, B.M. Metabolism of Nicotinamide Mononucleotide in Beef Liver. Arch. Biochem. Biophys. 1987, 254, 241–252. [Google Scholar] [CrossRef]
- Orecchia, A.; Scarponi, C.; Di Felice, F.; Cesarini, E.; Avitabile, S.; Mai, A.; Mauro, M.L.; Sirri, V.; Zambruno, G.; Albanesi, C.; et al. Sirtinol Treatment Reduces Inflammation in Human Dermal Microvascular Endothelial Cells. PLoS ONE 2011, 6, e24307. [Google Scholar] [CrossRef]
- She, D.T.; Wong, L.J.; Baik, S.-H.; Arumugam, T.V. SIRT2 Inhibition Confers Neuroprotection by Downregulation of FOXO3a and MAPK Signaling Pathways in Ischemic Stroke. Mol. Neurobiol. 2018, 55, 9188–9203. [Google Scholar] [CrossRef]
- Lee, A.S.; Jung, Y.J.; Kim, D.; Nguyen-Thanh, T.; Kang, K.P.; Lee, S.; Park, S.K.; Kim, W. SIRT2 Ameliorates Lipopolysaccharide-Induced Inflammation in Macrophages. Biochem. Biophys. Res. Commun. 2014, 450, 1363–1369. [Google Scholar] [CrossRef]
- Sabio, G.; Cavanagh-Kyros, J.; Ko, H.J.; Jung, D.Y.; Gray, S.; Jun, J.Y.; Barrett, T.; Mora, A.; Kim, J.K.; Davis, R.J. Prevention of Steatosis by Hepatic JNK1. Cell Metab. 2009, 10, 491–498. [Google Scholar] [CrossRef]
- Wrede, C.E.; Dickson, L.M.; Lingohr, M.K.; Briaud, I.; Rhodes, C.J. Protein Kinase B/Akt Prevents Fatty Acid-Induced Apoptosis in Pancreatic Beta-Cells (INS-1). J. Biol. Chem. 2002, 277, 49676–49684. [Google Scholar] [CrossRef]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H.; Wright, C.V.E.; White, M.F.; Arden, K.C.; Accili, D. The Forkhead Transcription Factor Foxo1 Links Insulin Signaling to Pdx1 Regulation of Pancreatic β Cell Growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef]
- Lee, J.-H.; Song, M.-Y.; Song, E.-K.; Kim, E.-K.; Moon, W.S.; Han, M.-K.; Park, J.-W.; Kwon, K.-B.; Park, B.-H. Overexpression of SIRT1 Protects Pancreatic β-Cells against Cytokine Toxicity by Suppressing the Nuclear Factor-KB Signaling Pathway. Diabetes 2009, 58, 344–351. [Google Scholar] [CrossRef]
- Ramsey, K.M.; Mills, K.F.; Satoh, A.; Imai, S.-I. Age-Associated Loss of Sirt1-Mediated Enhancement of Glucose-Stimulated Insulin Secretion in Beta Cell-Specific Sirt1-Overexpressing (BESTO) Mice. Aging Cell 2008, 7, 78–88. [Google Scholar] [CrossRef]
- Sandler, S.; Welsh, M.; Andersson, A. Streptozotocin-Induced Impairment of Islet B-Cell Metabolism and Its Prevention by a Hydroxyl Radical Scavenger and Inhibitors of Poly (ADP-Ribose) Synthetase. Acta Pharmacol. Toxicol. 1983, 53, 392–400. [Google Scholar] [CrossRef]
- Novelli, M.; Canistro, D.; Martano, M.; Funel, N.; Sapone, A.; Melega, S.; Masini, M.; De Tata, V.; Pippa, A.; Vecoli, C.; et al. Anti-Diabetic Properties of a Non-Conventional Radical Scavenger, as Compared to Pioglitazone and Exendin-4, in Streptozotocin-Nicotinamide Diabetic Mice. Eur. J. Pharmacol. 2014, 729, 37–44. [Google Scholar] [CrossRef]
- Sandler, S.; Andersson, A. Long-Term Effects of Exposure of Pancreatic Islets to Nicotinamide In Vitro on DNA Synthesis, Metabolism and B-Cell Function. Diabetologia 1986, 29, 199–202. [Google Scholar] [CrossRef]
- Sjöholm, A. Effects of Nicotinamide on Clonal Rat Insulinoma Cell Proliferation, Polyamine Content and Insulin Secretion. Anticancer Res. 1993, 13, 1283–1285. [Google Scholar] [PubMed]
- Watanabe, H.; Inaba, Y.; Kimura, K.; Matsumoto, M.; Kaneko, S.; Kasuga, M.; Inoue, H. Sirt2 Facilitates Hepatic Glucose Uptake by Deacetylating Glucokinase Regulatory Protein. Nat. Commun. 2018, 9, 30. [Google Scholar] [CrossRef]
- Wang, R.-H.; Xu, X.; Kim, H.-S.; Xiao, Z.; Deng, C.-X. SIRT1 Deacetylates FOXA2 and Is Critical for Pdx1 Transcription and β-Cell Formation. Int. J. Biol. Sci. 2013, 9, 934–946. [Google Scholar] [CrossRef]
- Lantier, L.; Williams, A.S.; Hughey, C.C.; Bracy, D.P.; James, F.D.; Ansari, M.A.; Gius, D.; Wasserman, D.H. SIRT2 Knockout Exacerbates Insulin Resistance in High Fat-Fed Mice. PLoS ONE 2018, 13, e0208634. [Google Scholar] [CrossRef]
- DeAngelis, A.M.; Heinrich, G.; Dai, T.; Bowman, T.A.; Patel, P.R.; Lee, S.J.; Hong, E.-G.; Jung, D.Y.; Assmann, A.; Kulkarni, R.N.; et al. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1: A Link Between Insulin and Lipid Metabolism. Diabetes 2008, 57, 2296–2303. [Google Scholar] [CrossRef]
- Xu, E.; Dubois, M.-J.; Leung, N.; Charbonneau, A.; Turbide, C.; Avramoglu, R.K.; DeMarte, L.; Elchebly, M.; Streichert, T.; Lévy, E.; et al. Targeted Disruption of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Promotes Diet-Induced Hepatic Steatosis and Insulin Resistance. Endocrinology 2009, 150, 3503–3512. [Google Scholar] [CrossRef] [PubMed]
- Berzins, R.; Wieczorek, K.R.; Rajotte, R.V.; Molnar, G.D.; Tam, Y.K.; McGregor, J.R.; Fawcett, D.M. Accuracy of C-Peptide:Insulin Molar Ratio as a Measure of Hepatic Removal of Insulin. Diabetes Res. Clin. Pract. 1987, 4, 37–43. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of Oral Administration of Nicotinamide Mononucleotide on Clinical Parameters and Nicotinamide Metabolite Levels in Healthy Japanese Men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef]
- Bezman-Tarcher, A. Method for Continuous Intravenous Infusion of Large Amounts of Oleic Acid into Rats. J. Lipid Res. 1969, 10, 197–206. [Google Scholar] [CrossRef]
- Basu, A.; Basu, R.; Shah, P.; Vella, A.; Rizza, R.A.; Jensen, M.D. Systemic and Regional Free Fatty Acid Metabolism in Type 2 Diabetes. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E1000–E1006. [Google Scholar] [CrossRef]
- Tang, C.; Han, P.; Oprescu, A.I.; Lee, S.C.; Gyulkhandanyan, A.V.; Chan, G.N.Y.; Wheeler, M.B.; Giacca, A. Evidence for a Role of Superoxide Generation in Glucose-Induced Beta-Cell Dysfunction In Vivo. Diabetes 2007, 56, 2722–2731. [Google Scholar] [CrossRef] [PubMed][Green Version]
- DeFronzo, R.A.; Tobin, J.D.; Andres, R. Glucose Clamp Technique: A Method for Quantifying Insulin Secretion and Resistance. Am. J. Physiol. 1979, 237, E214–E223. [Google Scholar] [CrossRef] [PubMed]
- Natali, A.; Gastaldelli, A.; Camastra, S.; Sironi, A.M.; Toschi, E.; Masoni, A.; Ferrannini, E.; Mari, A. Dose-Response Characteristics of Insulin Action on Glucose Metabolism: A Non-Steady-State Approach. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E794–E801. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Naassan, A.E.; Chamson-Reig, A.; Koulajian, K.; Goh, T.T.; Yoon, F.; Oprescu, A.I.; Ghanim, H.; Lewis, G.F.; Dandona, P.; et al. Susceptibility to Fatty Acid-Induced β-Cell Dysfunction Is Enhanced in Prediabetic Diabetes-Prone BioBreeding Rats: A Potential Link Between β-Cell Lipotoxicity and Islet Inflammation. Endocrinology 2013, 154, 89–101. [Google Scholar] [CrossRef]
- Goh, T.T.; Mason, T.M.; Gupta, N.; So, A.; Lam, T.K.T.; Lam, L.; Lewis, G.F.; Mari, A.; Giacca, A. Lipid-Induced β-Cell Dysfunction In Vivo in Models of Progressive β-Cell Failure. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E549–E560. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y.; Yoshida, A.; Suganami, H.; Ishiguro, H.; Yamamoto, M.; Fujihara, K.; Kodama, S.; Tanaka, S.; Kaku, K.; Sone, H. Role of Fatty Liver in the Association between Obesity and Reduced Hepatic Insulin Clearance. Diabetes Metab. 2018, 44, 135–142. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||
|---|---|---|
| Ad-Control | Ad-Sirt1 | |
| Sirt1 mRNA | <0.01 | 0.63 ± 0.07 ** |
| Ceacam1 mRNA | 1.66 ± 0.19 | 1.47 ± 0.21 |
| (B) | ||
| Ad-Control | Ad-Sirt2 | |
| Sirt2 mRNA | 1.03 ± 0.02 | 20.97 ± 3.62 ** |
| Ceacam1 mRNA | 0.99 ± 0.03 | 1.81 ± 0.07 * |
| (C) | ||
| Ad-Control | Ad-Control + Sirtinol | |
| Sirt2 mRNA | 1.11 ± 0.05 | 1.25 ± 0.20 |
| Ceacam1 mRNA | 1.02 ± 0.03 | 0.35 ± 0.04 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nahle, A.; Joseph, Y.D.; Pereira, S.; Mori, Y.; Poon, F.; Ghadieh, H.E.; Ivovic, A.; Desai, T.; Ghanem, S.S.; Asalla, S.; et al. Nicotinamide Mononucleotide Prevents Free Fatty Acid-Induced Reduction in Glucose Tolerance by Decreasing Insulin Clearance. Int. J. Mol. Sci. 2021, 22, 13224. https://doi.org/10.3390/ijms222413224
Nahle A, Joseph YD, Pereira S, Mori Y, Poon F, Ghadieh HE, Ivovic A, Desai T, Ghanem SS, Asalla S, et al. Nicotinamide Mononucleotide Prevents Free Fatty Acid-Induced Reduction in Glucose Tolerance by Decreasing Insulin Clearance. International Journal of Molecular Sciences. 2021; 22(24):13224. https://doi.org/10.3390/ijms222413224
Chicago/Turabian StyleNahle, Ashraf, Yemisi Deborah Joseph, Sandra Pereira, Yusaku Mori, Frankie Poon, Hilda E. Ghadieh, Aleksandar Ivovic, Tejas Desai, Simona S. Ghanem, Suman Asalla, and et al. 2021. "Nicotinamide Mononucleotide Prevents Free Fatty Acid-Induced Reduction in Glucose Tolerance by Decreasing Insulin Clearance" International Journal of Molecular Sciences 22, no. 24: 13224. https://doi.org/10.3390/ijms222413224
APA StyleNahle, A., Joseph, Y. D., Pereira, S., Mori, Y., Poon, F., Ghadieh, H. E., Ivovic, A., Desai, T., Ghanem, S. S., Asalla, S., Muturi, H. T., Jentz, E. M., Joseph, J. W., Najjar, S. M., & Giacca, A. (2021). Nicotinamide Mononucleotide Prevents Free Fatty Acid-Induced Reduction in Glucose Tolerance by Decreasing Insulin Clearance. International Journal of Molecular Sciences, 22(24), 13224. https://doi.org/10.3390/ijms222413224