
Novel Strategy of Proxalutamide for the Treatment of Prostate Cancer through Coordinated Blockade of Lipogenesis and Androgen Receptor Axis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
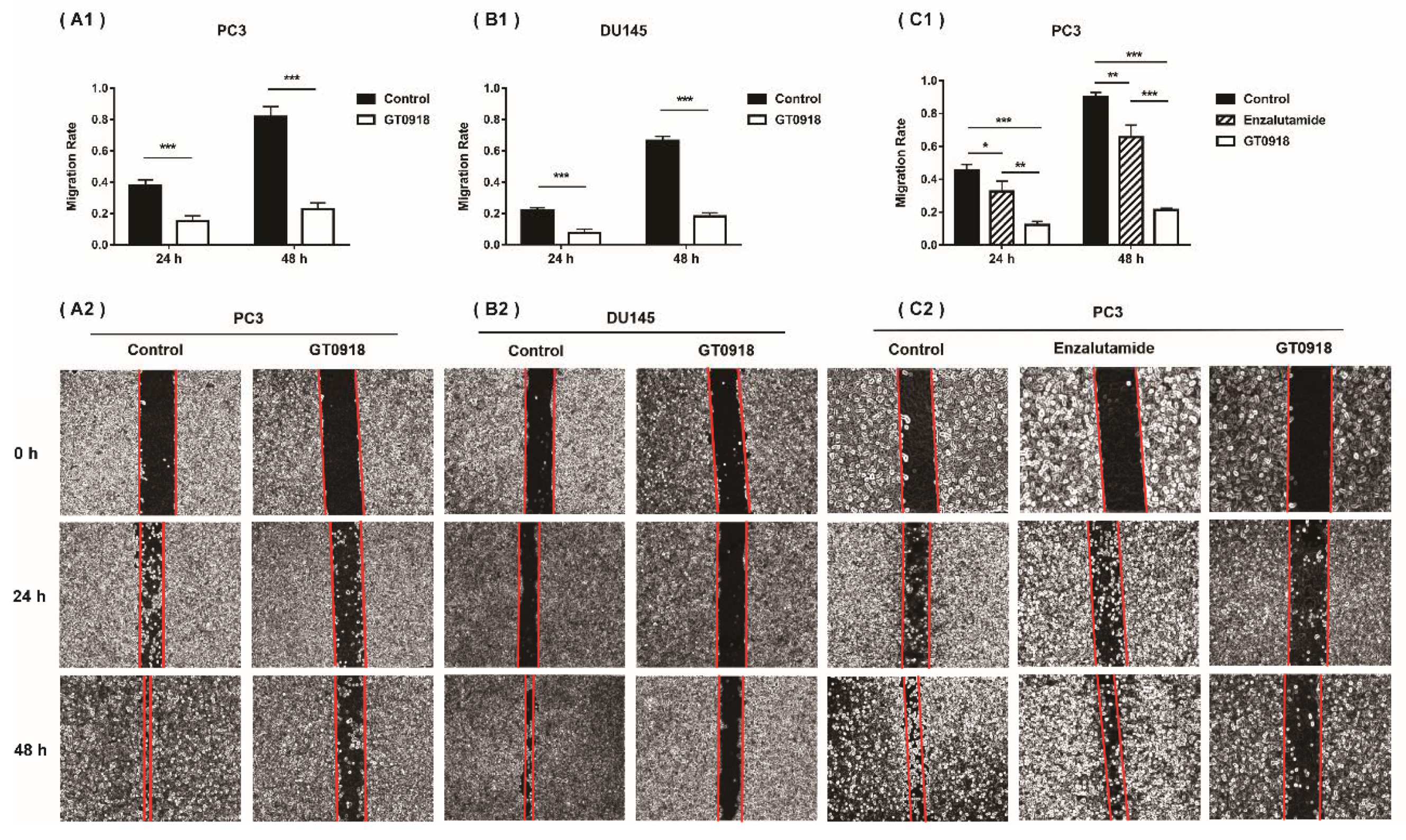
2.1. Proxalutamide Suppresses PCa Cell Growth and Migration
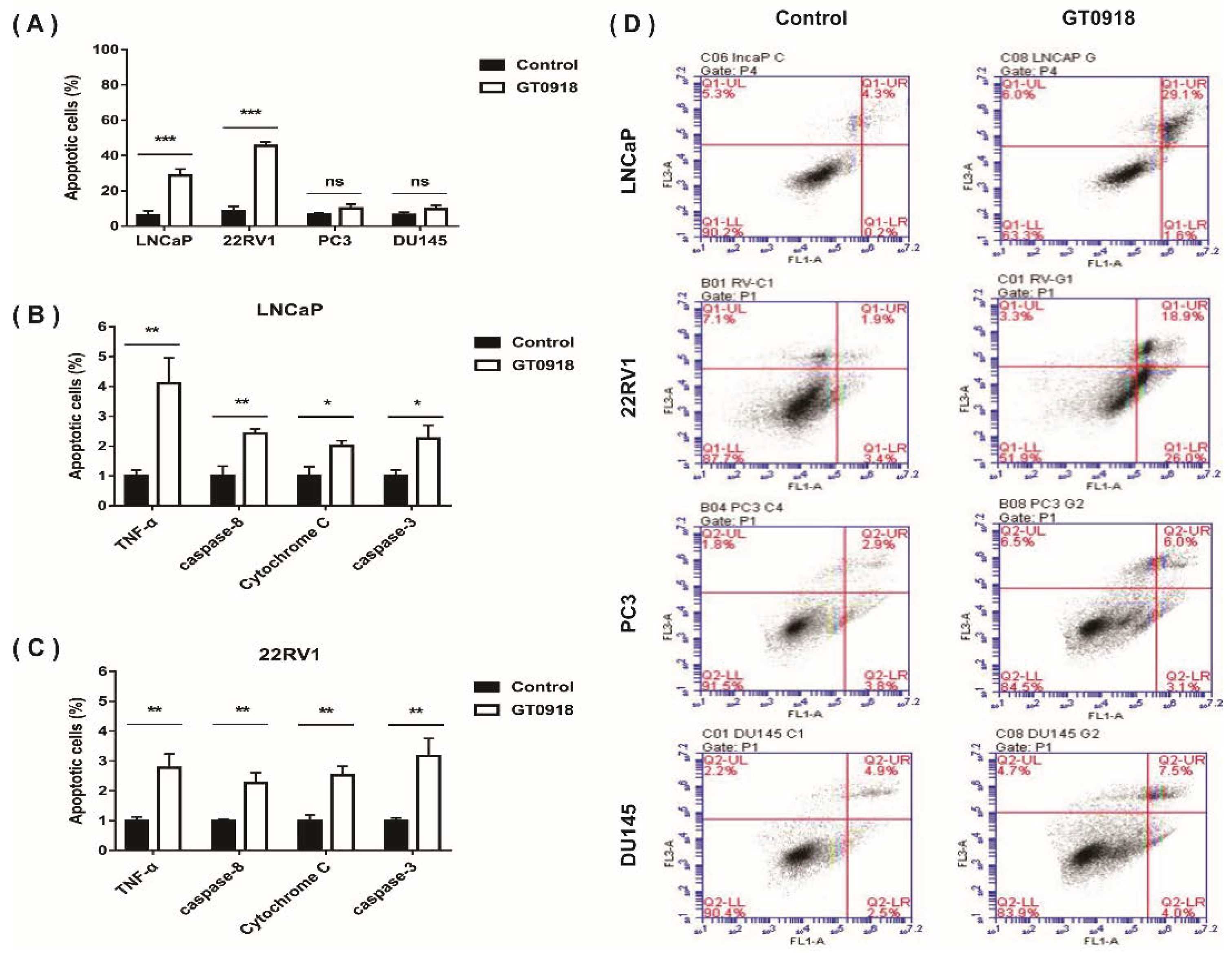
2.2. Proxalutamide Induces Caspase-Dependent Apoptosis Leading to PCa Cell Death
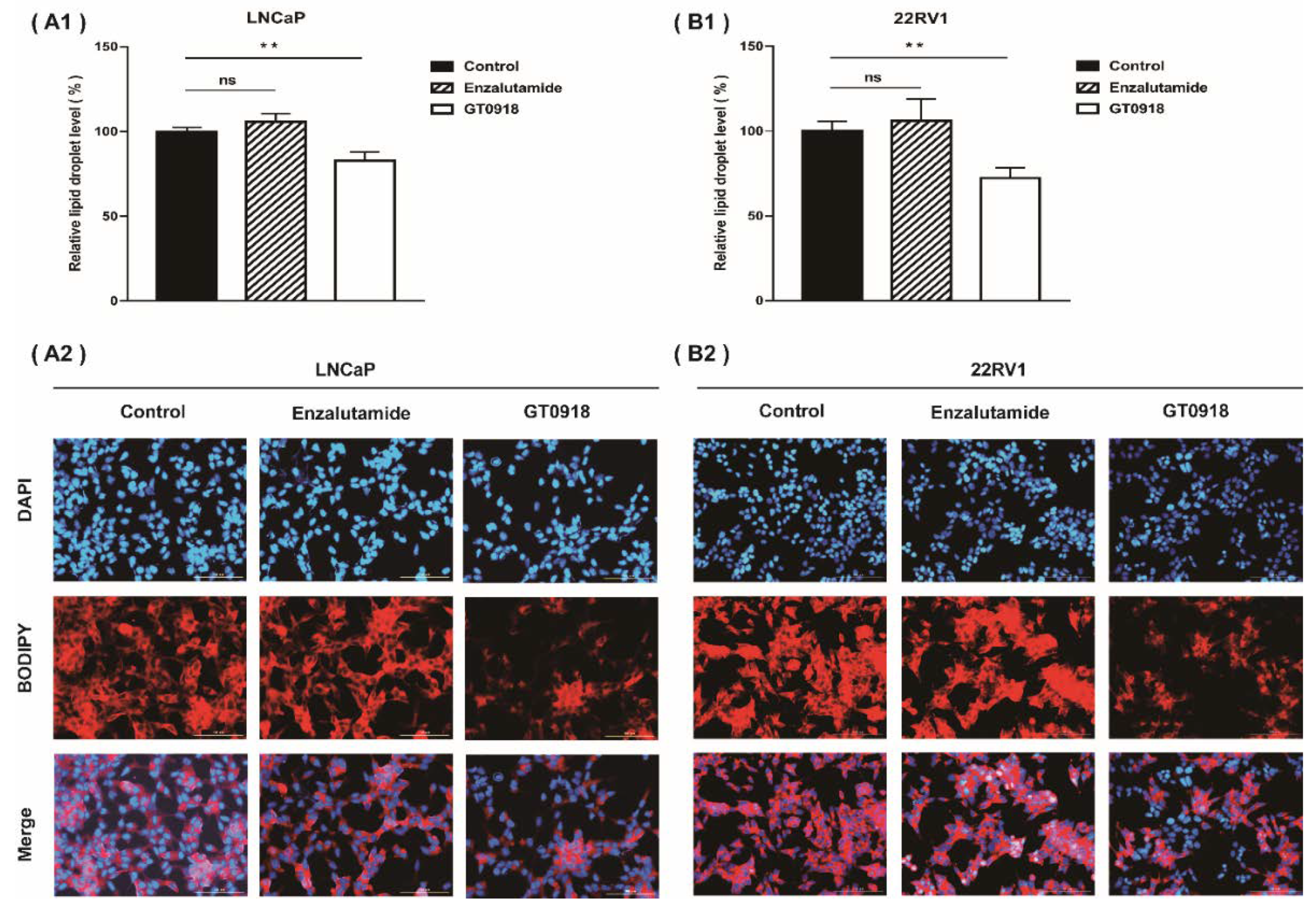
2.3. Proxalutamide Reduces Lipid Enrichment in PCa Cells
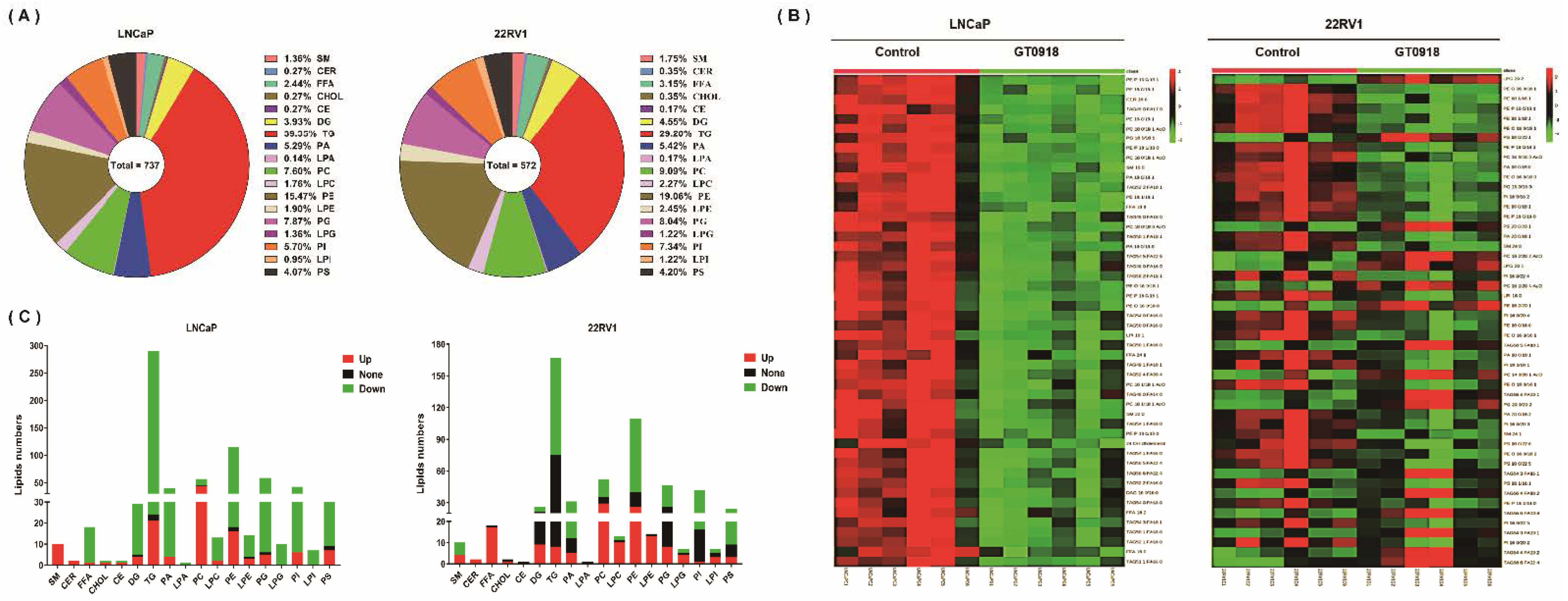
2.4. Proxalutamide Changes Lipid Profile of PCa Cells
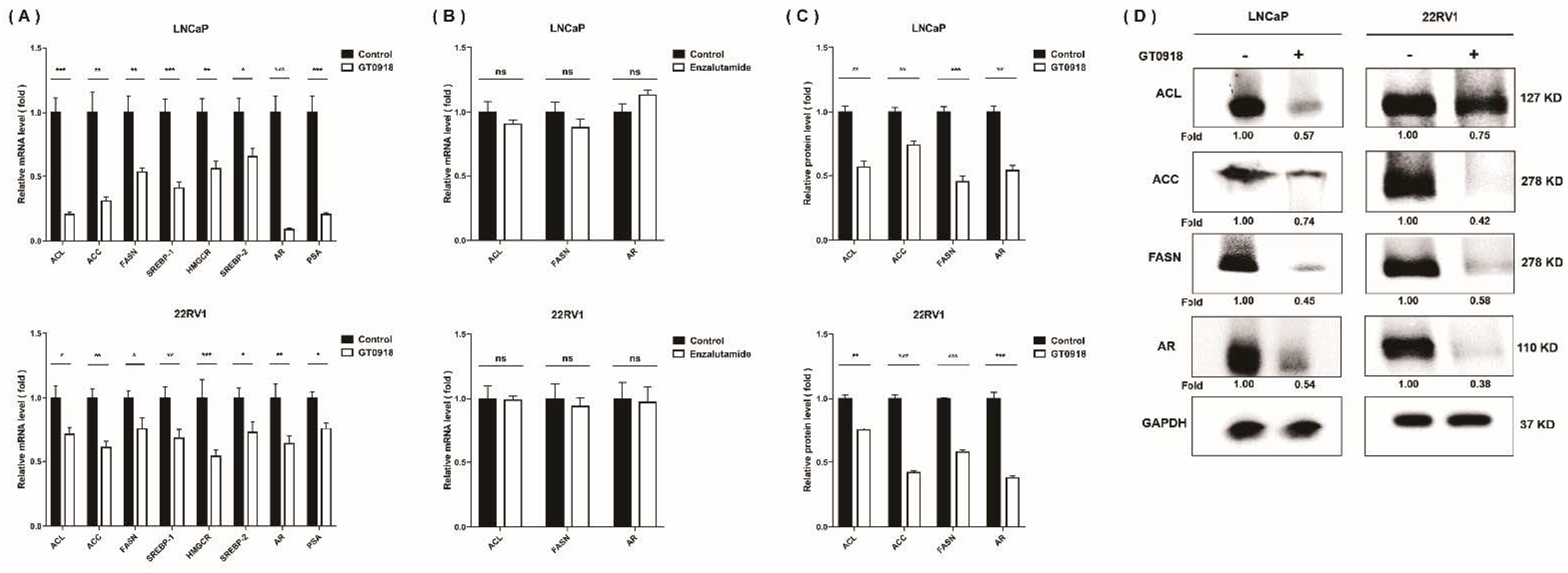
2.5. Proxalutamide Deceases Lipogenesis in PCa Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Analysis of PCa Cell Migration
4.4. Analysis of PCa Cell Apoptosis
4.5. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-qPCR)
4.6. Western Blotting
4.7. Detection of LDs by Fluorescent Microscopy
4.8. Lipidomics Analysis
4.8.1. Lipidomics Sample Preparation
4.8.2. Lipidomics Sample Detection
4.8.3. Lipidomics Data Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Thin, R.N. Prostatitis. Hosp. Med. 1999, 60, 710–713. [Google Scholar] [CrossRef]
- Langan, R.C. Benign Prostatic Hyperplasia. Prim. Care 2019, 46, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Nielsen, M.; Borre, M. Diagnostic and Therapeutic Strategies for Prostate Cancer. Semin. Nucl. Med. 2016, 46, 484–490. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J.; Sharifi, N. Sex Hormones and Prostate Cancer. Annu. Rev. Med. 2020, 71, 33–45. [Google Scholar] [CrossRef]
- Ritch, C.; Cookson, M. Recent trends in the management of advanced prostate cancer. F1000Research 2018, 7, 1513. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. Expert Opin. Pharmacother. 2008, 9, 211–228. [Google Scholar] [CrossRef]
- Mizokami, A.; Namiki, M. Reconsideration of progression to CRPC during androgen deprivation therapy. J. Steroid Biochem. Mol. Biol. 2015, 145, 164–171. [Google Scholar] [CrossRef]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Dellis, A.E.; Papatsoris, A.G. Perspectives on the current and emerging chemical androgen receptor antagonists for the treatment of prostate cancer. Expert Opin. Pharmacother. 2018, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S. Progress in emerging therapies for advanced prostate cancer. Cancer Treat. Rev. 2013, 39, 275–289. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2010, 8, 12–23. [Google Scholar] [CrossRef]
- Pomerantz, M.; Kantoff, P. Advances in the Treatment of Prostate Cancer. Annu. Rev. Med. 2007, 58, 205–220. [Google Scholar] [CrossRef]
- Zhou, T.; Xu, W.; Zhang, W.; Sun, Y.; Yan, H.; Gao, X.; Wang, F.; Zhou, Q.; Hou, J.; Ren, S.; et al. Preclinical profile and phase I clinical trial of a novel androgen receptor antagonist GT0918 in castration-resistant prostate cancer. Eur. J. Cancer 2020, 134, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Chen, C.; Wu, J.; Yang, J.; Zhang, H.; Wu, X.; Duan, Y.; Gao, W.; Qian, W.; Niu, X.; et al. Abstract 614: Proxalutamide (GT0918), a potent androgen receptor pathway inhibitor. Endocrinology 2014, 74, 614. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Qu, F.; Gu, Y.; Wang, Q.; He, M.; Zhou, F.; Sun, J.; Wang, G.; Peng, Y. Metabolomic profiling to evaluate the efficacy of proxalutamide, a novel androgen receptor antagonist, in prostate cancer cells. Investig. New Drugs 2020, 38, 1292–1302. [Google Scholar] [CrossRef]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Griffin, J.E. Androgen Resistance—The Clinical and Molecular Spectrum. N. Engl. J. Med. 1992, 326, 611–618. [Google Scholar] [CrossRef]
- Singh, K.B.; Singh, S.V. Fatty Acid Synthesis Intermediates Represent Novel Noninvasive Biomarkers of Prostate Cancer Chemoprevention by Phenethyl Isothiocyanate. Cancer Prev. Res. 2017, 10, 279–289. [Google Scholar] [CrossRef]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.; Machiels, J.; et al. De novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef]
- Tousignant, K.; Rockstroh, A.; Fard, A.T.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol. Cancer Res. 2019, 17, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Gang, X.; Yang, Y.; Zhong, J.; Jiang, K.; Pan, Y.; Karnes, R.J.; Zhang, J.; Xu, W.; Wang, G.; Huang, H. P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget 2016, 7, 15135–15149. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: Evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef]
- Balusamy, S.R.; Perumalsamy, H.; Veerappan, K.; Huq, A.; RajeshKumar, S.; Lakshmi, T.; Kim, Y.J. Citral Induced Apoptosis through Modulation of Key Genes Involved in Fatty Acid Biosynthesis in Human Prostate Cancer Cells: In Silico and In Vitro Study. BioMed Res. Int. 2020, 2020, 6040727. [Google Scholar] [CrossRef]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Futur. Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef]
- Chuang, H.Y.; Lee, Y.P.; Lin, W.C.; Lin, Y.H.; Hwang, J.J. Fatty Acid Inhibition Sensitizes Androgen-Dependent and -Independent Prostate Cancer to Radiotherapy via FASN/NF-kappaB Pathway. Sci. Rep. 2019, 9, 13284. [Google Scholar] [CrossRef]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.F.; Jiang, W.P.; Huang, S.Y.; Basavaraj, P.; Wu, J.B.; Ho, H.Y.; Huang, G.J.; Huang, W.C. Davallia formosanaEmerging Therapeutic Activity of on Prostate Cancer Cells through Coordinated Blockade of Lipogenesis and Androgen Receptor Expression. Cancers 2020, 12, 914. [Google Scholar] [CrossRef]
- Singh, K.B.; Kim, S.-H.; Hahm, E.-R.; Pore, S.K.; Jacobs, B.L.; Singh, S.V. Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis 2018, 39, 826–837. [Google Scholar] [CrossRef]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Ito, M.; Minamiya, Y.; Kawai, H.; Ogawa, J. P-056 Primary tumor-derived TGF-B1 induces dendritic cell apoptosisin sentinel lymph nodes of patients with non-small cell lung cancer. Lung Cancer 2005, 49, S129. [Google Scholar] [CrossRef]
- Huang, S.; Huang, G.; Hsieh, P.; Wu, H.; Huang, W. Osajin displays potential antiprostate cancer efficacy via impairment of fatty acid synthase and androgen receptor expression. Prostate 2019, 79, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Huang, G.J.; Wu, H.C.; Kao, M.C.; Huang, W.C. Ganoderma tsugae Inhibits the SREBP-1/AR Axis Leading to Suppression of Cell Growth and Activation of Apoptosis in Prostate Cancer Cells. Molecules 2018, 23, 2539. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.-T.; Hu, P.; Huang, W.-C. Fatostatin Displays High Antitumor Activity in Prostate Cancer by Blocking SREBP-Regulated Metabolic Pathways and Androgen Receptor Signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Hermel, E.; Castro-Obregon, S.; del Rio, G.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling Endoplasmic Reticulum Stress to the Cell Death Program. J. Biol. Chem. 2001, 276, 33869–33874. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- Deep, G.; Schlaepfer, I.R. Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. Int. J. Mol. Sci. 2016, 17, 1061. [Google Scholar] [CrossRef]
- Torimoto, K.; Samma, S.; Kagebayashi, Y.; Chihara, Y.; Tanaka, N.; Hirayama, A.; Fujimoto, K.; Hirao, Y. The Effects of Androgen Deprivation Therapy on Lipid Metabolism and Body Composition in Japanese Patients with Prostate Cancer. Jpn. J. Clin. Oncol. 2011, 41, 577–581. [Google Scholar] [CrossRef][Green Version]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2013, 1831, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of Sterol Response Element-Binding Proteins and Downstream Effectors in Prostate Cancer during Progression to Androgen Independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef]
- Rossi, S.; Graner, E.; Febbo, P.; Weinstein, L.; Bhattacharya, N.; Onody, T.; Bubley, G.; Balk, S.; Loda, M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer Res. 2003, 1, 707–715. [Google Scholar] [PubMed]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Teoh, J.Y.C.; Hirai, H.W.; Ho, J.M.W.; Chan, F.C.H.; Tsoi, K.K.F.; Ng, C.F. Global incidence of prostate cancer in developing and developed countries with changing age structures. PLoS ONE 2019, 14, e0221775. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef]
- Jenster, G. The role of the androgen receptor in the development and progression of prostate cancer. Semin. Oncol. 1999, 26, 407–421. [Google Scholar]
- Dutt, S.S.; Gao, A.C. Molecular mechanisms of castration-resistant prostate cancer progression. Futur. Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B. Enzalutamide and Cancer. 2018
- Obinata, D.; Lawrence, M.G.; Takayama, K.; Choo, N.; Risbridger, G.P.; Takahashi, S.; Inoue, S. Recent Discoveries in the Androgen Receptor Pathway in Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 581515. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Yuan, X.; Cai, C.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2013, 33, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Spisák, S.; Seo, J.-H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Armstrong, A.J.; Dehm, S.M.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen Receptor Variants Occur Frequently in Castration Resistant Prostate Cancer Metastases. PLoS ONE 2011, 6, e27970. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Sarwar, M.; Semenas, J.; Miftakhova, R.; Simoulis, A.; Robinson, B.; Wingren, A.G.; Mongan, N.; Heery, D.; Johnson, H.; Abrahamsson, P.-A.; et al. Targeted suppression of AR-V7 using PIP5K1α inhibitor overcomes enzalutamide resistance in prostate cancer cells. Oncotarget 2016, 7, 63065–63081. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Wu, Z.; Ding, W.; Gao, S.; Gao, Y.; Xu, C. Mechanisms of enzalutamide resistance in castration-resistant prostate cancer and therapeutic strategies to overcome it. Br. J. Pharmacol. 2020, 178, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Lounis, M.A.; Péant, B.; Leclerc-Desaulniers, K.; Ganguli, D.; Daneault, C.; Ruiz, M.; Zoubeidi, A.; Mes-Masson, A.-M.; Saad, F. Modulation of de Novo Lipogenesis Improves Response to Enzalutamide Treatment in Prostate Cancer. Cancers 2020, 12, 3339. [Google Scholar] [CrossRef]
- Kong, Y.; Cheng, L.; Mao, F.; Zhang, Z.; Zhang, Y.; Farah, E.; Bosler, J.; Bai, Y.; Ahmad, N.; Kuang, S.; et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 2018, 293, 14328–14341. [Google Scholar] [CrossRef]
- Massie, C.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Krongrad, A.; Wilson, C.M.; Wilson, J.D.; Allman, D.R.; McPhaul, M.J. Androgen increases androgen receptor protein while decreasing receptor mRNA in LNCaP cells. Mol. Cell. Endocrinol. 1991, 76, 79–88. [Google Scholar] [CrossRef]
- Komendantova, A.S.; Scherbakov, A.M.; Komkov, A.V.; Chertkova, V.V.; Gudovanniy, A.O.; Chernoburova, E.I.; Sorokin, D.V.; Dzichenka, Y.; Shirinian, V.; Volkova, Y.A.; et al. Novel steroidal 1,3,4-thiadiazines: Synthesis and biological evaluation in androgen receptor-positive prostate cancer 22Rv1 cells. Bioorg. Chem. 2019, 91, 103142. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line--a new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.-S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. Vitr. Cell. Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Rahimi, S.; Roushandeh, A.M.; Ebrahimi, A.; Samadani, A.A.; Kuwahara, Y.; Roudkenar, M.H. CRISPR/Cas9-mediated knockout of Lcn2 effectively enhanced CDDP-induced apoptosis and reduced cell migration capacity of PC3 cells. Life Sci. 2019, 231, 116586. [Google Scholar] [CrossRef]
- Chen, J.; Wang, F.; Xu, H.; Chen, D.; Liu, W.; Wang, J. Over-expression of TM4SF1 improves cell metastasis and growth by activating ERK1/2 signaling pathway in human prostate cancer. J. BUON 2019, 24, 2531–2538. [Google Scholar]
- Rudzinski, J.K.; Govindasamy, N.P.; Lewis, J.D.; Jurasz, P. The role of the androgen receptor in prostate cancer-induced platelet aggregation and platelet-induced invasion. J. Thromb. Haemost. 2020, 18, 2976–2986. [Google Scholar] [CrossRef]
- Kan, S.-F.; Yu, C.-H.; Pu, H.-F.; Hsu, J.-M.; Chen, M.-J.; Wang, P.S. Anti-proliferative effects of evodiamine on human prostate cancer cell lines DU145 and PC3. J. Cell. Biochem. 2007, 101, 44–56. [Google Scholar] [CrossRef]
- Fiorentini, C.; Bodei, S.; Bedussi, F.; Fragni, M.; Bonini, S.; Simeone, C.; Zani, D.; Berruti, A.; Missale, C.; Memo, M.; et al. GPNMB/OA protein increases the invasiveness of human metastatic prostate cancer cell lines DU145 and PC3 through MMP-2 and MMP-9 activity. Exp. Cell Res. 2014, 323, 100–111. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Xue, M.; Wang, Q.; Hong, X.; Wang, X.; Zhou, F.; Sun, J.; Wang, G.; Peng, Y. Novel Strategy of Proxalutamide for the Treatment of Prostate Cancer through Coordinated Blockade of Lipogenesis and Androgen Receptor Axis. Int. J. Mol. Sci. 2021, 22, 13222. https://doi.org/10.3390/ijms222413222
Gu Y, Xue M, Wang Q, Hong X, Wang X, Zhou F, Sun J, Wang G, Peng Y. Novel Strategy of Proxalutamide for the Treatment of Prostate Cancer through Coordinated Blockade of Lipogenesis and Androgen Receptor Axis. International Journal of Molecular Sciences. 2021; 22(24):13222. https://doi.org/10.3390/ijms222413222
Chicago/Turabian StyleGu, Yue, Mengxia Xue, Qizhi Wang, Xiaodan Hong, Xinyu Wang, Fang Zhou, Jianguo Sun, Guangji Wang, and Ying Peng. 2021. "Novel Strategy of Proxalutamide for the Treatment of Prostate Cancer through Coordinated Blockade of Lipogenesis and Androgen Receptor Axis" International Journal of Molecular Sciences 22, no. 24: 13222. https://doi.org/10.3390/ijms222413222
APA StyleGu, Y., Xue, M., Wang, Q., Hong, X., Wang, X., Zhou, F., Sun, J., Wang, G., & Peng, Y. (2021). Novel Strategy of Proxalutamide for the Treatment of Prostate Cancer through Coordinated Blockade of Lipogenesis and Androgen Receptor Axis. International Journal of Molecular Sciences, 22(24), 13222. https://doi.org/10.3390/ijms222413222