
eIF5A-Independent Role of DHPS in p21CIP1 and Cell Fate Regulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DHPS Depletion Increases p21CIP1 Levels and Induces G0/G1 Phase Cell Cycle Arrest in A375 and SK-MEL-28 Cells
2.2. eIF5A Depletion Induces Cell Death without Increasing p21CIP1 Levels in A375 and SK-MEL-28 Cells
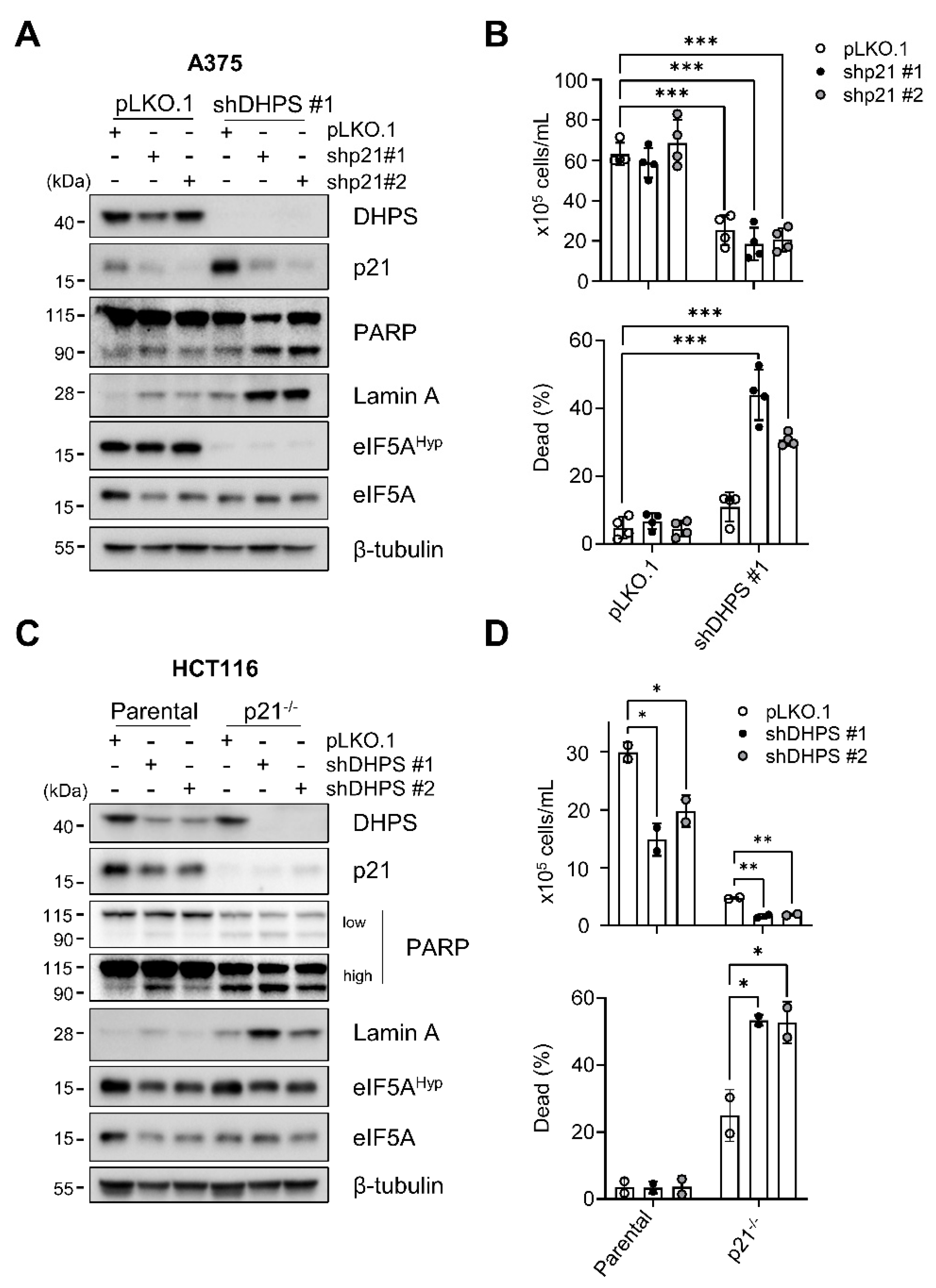
2.3. p21CIP1 Depletion Switches DHPS Depletion-Induced Growth Arrest to Cell Death
2.4. Hypusinating Activity Is Not Necessary for DHPS to Regulate p21CIP1
2.5. MEK/ERK Activity Is Necessary for DHPS Knockdown to Increase p21CIP1 Levels in Cells
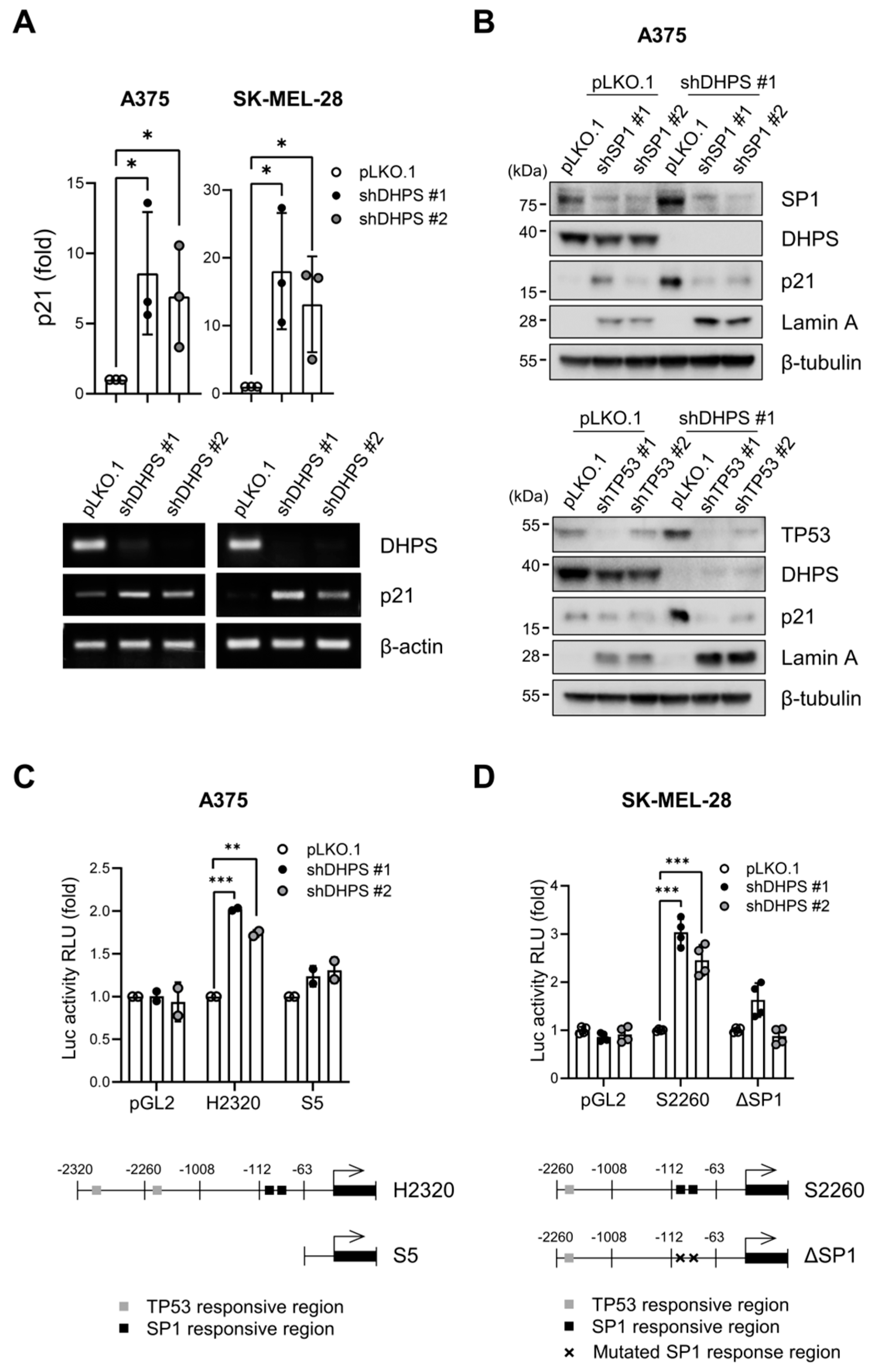
2.6. DHPS Knockdown Increases CDKN1A Transcription via TP53 and SP1 in A375 and SK-MEL-28 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Plasmids and Viral Constructs for Gene Expression and RNA Interference
4.3. Analysis of Cell Viability and Cell Cycle
4.4. Quantitative RT-PCR (qPCR) and Luciferase Reporter Assays
4.5. Immunoblotting
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joe, Y.A.; Wolff, E.C.; Park, M.H. Cloning and expression of human deoxyhypusine synthase cDNA. Structure-function studies with the recombinant enzyme and mutant proteins. J. Biol. Chem. 1995, 270, 22386–22392. [Google Scholar] [CrossRef]
- Wolff, E.C.; Lee, Y.B.; Chung, S.I.; Folk, J.E.; Park, M.H. Deoxyhypusine synthase from rat testis: Purification and characterization. J. Biol. Chem. 1995, 270, 8660–8666. [Google Scholar] [CrossRef] [PubMed]
- Su, W.Y.; Li, J.T.; Cui, Y.; Hong, J.; Du, W.; Wang, Y.C.; Lin, Y.W.; Xiong, H.; Wang, J.L.; Kong, X.; et al. Bidirectional regulation between WDR83 and its natural antisense transcript DHPS in gastric cancer. Cell Res. 2012, 22, 1374–1389. [Google Scholar] [CrossRef][Green Version]
- Park, M.H. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A). J. Biochem. 2006, 139, 161–169. [Google Scholar] [CrossRef]
- Park, M.H.; Wolff, E.C. Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 2018, 293, 18710–18718. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.L.; Park, M.H.; Folk, J.E.; Safer, B.; Braverman, R. Identification of the hypusine-containing protein hy+ as translation initiation factor eIF-4D. Proc. Natl. Acad. Sci. USA 1983, 80, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gai, Z.; Okada, C.; Ye, Y.; Yu, J.; Yao, M. Flexible NAD(+) Binding in Deoxyhypusine Synthase Reflects the Dynamic Hypusine Modification of Translation Factor IF5A. Int. J. Mol. Sci. 2020, 21, 5509. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Um, P.Y.; Park, M.H. Structure-function studies of human deoxyhypusine synthase: Identification of amino acid residues critical for the binding of spermidine and NAD. Biochem. J. 2001, 355, 841–849. [Google Scholar] [CrossRef]
- Umland, T.C.; Wolff, E.C.; Park, M.H.; Davies, D.R. A new crystal structure of deoxyhypusine synthase reveals the configuration of the active enzyme and of an enzyme.NAD.inhibitor ternary complex. J. Biol. Chem. 2004, 279, 28697–28705. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kang, K.R.; Wolff, E.C.; Bell, J.K.; McPhie, P.; Park, M.H. Deoxyhypusine hydroxylase is a Fe(II)-dependent, HEAT-repeat enzyme. Identification of amino acid residues critical for Fe(II) binding and catalysis [corrected]. J. Biol. Chem. 2006, 281, 13217–13225. [Google Scholar] [CrossRef]
- Park, J.H.; Aravind, L.; Wolff, E.C.; Kaevel, J.; Kim, Y.S.; Park, M.H. Molecular cloning, expression, and structural prediction of deoxyhypusine hydroxylase: A HEAT-repeat-containing metalloenzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Q.; Xu, Y.M.; Lau, A.T.Y. Recent insights into eukaryotic translation initiation factors 5A1 and 5A2 and their roles in human health and disease. Cancer Cell Int. 2020, 20, 142. [Google Scholar] [CrossRef]
- Clement, P.M.; Johansson, H.E.; Wolff, E.C.; Park, M.H. Differential expression of eIF5A-1 and eIF5A-2 in human cancer cells. FEBS J. 2006, 273, 1102–1114. [Google Scholar] [CrossRef]
- Jenkins, Z.A.; Haag, P.G.; Johansson, H.E. Human eIF5A2 on chromosome 3q25-q27 is a phylogenetically conserved vertebrate variant of eukaryotic translation initiation factor 5A with tissue-specific expression. Genomics 2001, 71, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Alepuz, P. eIF5A facilitates translation termination globally and promotes the elongation of many non polyproline-specific tripeptide sequences. Nucleic Acids Res. 2017, 45, 7326–7338. [Google Scholar] [CrossRef]
- Schuller, A.P.; Wu, C.C.; Dever, T.E.; Buskirk, A.R.; Green, R. eIF5A Functions Globally in Translation Elongation and Termination. Mol. Cell 2017, 66, 194–205.e5. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.; Shin, B.S.; Woolstenhulme, C.J.; Kim, J.R.; Saini, P.; Buskirk, A.R.; Dever, T.E. eIF5A promotes translation of polyproline motifs. Mol. Cell 2013, 51, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Wolff, E.C.; Smit-McBride, Z.; Hershey, J.W.; Folk, J.E. Comparison of the activities of variant forms of eIF-4D. The requirement for hypusine or deoxyhypusine. J. Biol. Chem. 1991, 266, 7988–7994. [Google Scholar] [CrossRef]
- Park, M.H. The essential role of hypusine in eukaryotic translation initiation factor 4D (eIF-4D). Purification of eIF-4D and its precursors and comparison of their activities. J. Biol. Chem. 1989, 264, 18531–18535. [Google Scholar] [CrossRef]
- Schnier, J.; Schwelberger, H.G.; Smit-McBride, Z.; Kang, H.A.; Hershey, J.W. Translation initiation factor 5A and its hypusine modification are essential for cell viability in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 3105–3114. [Google Scholar] [CrossRef]
- Mathews, M.B.; Hershey, J.W. The translation factor eIF5A and human cancer. Biochim. Biophys. Acta 2015, 1849, 836–844. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Park, J.I. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front. Biol. 2014, 9, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Imperial, R.; Toor, O.M.; Hussain, A.; Subramanian, J.; Masood, A. Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: Its clinical implications. Semin. Cancer Biol. 2019, 54, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Bandino, A.; Geerts, D.; Koster, J.; Bachmann, A.S. Deoxyhypusine synthase (DHPS) inhibitor GC7 induces p21/Rb-mediated inhibition of tumor cell growth and DHPS expression correlates with poor prognosis in neuroblastoma patients. Cell Oncol. 2014, 37, 387–398. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Rao, L.; Perez, D.; White, E. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 1996, 135, 1441–1455. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol. Cell. Biol. 2003, 23, 543–554. [Google Scholar] [CrossRef]
- Hong, S.K.; Wu, P.K.; Park, J.I. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell. Signal. 2018, 42, 11–20. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, H.K.; Park, H.E.; Park, M.H.; Joe, Y.A. Effect of N1-guanyl-1,7-diaminoheptane, an inhibitor of deoxyhypusine synthase, on endothelial cell growth, differentiation and apoptosis. Mol. Cell. Biochem. 2002, 237, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.L.; Weber, M.J.; Bishop, J.M.; McMahon, M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human raf-1 protein kinase. Mol. Cell. Biol. 1993, 13, 6241–6252. [Google Scholar]
- Joe, Y.A.; Wolff, E.C.; Lee, Y.B.; Park, M.H. Enzyme-substrate intermediate at a specific lysine residue is required for deoxyhypusine synthesis. The role of Lys329 in human deoxyhypusine synthase. J. Biol. Chem. 1997, 272, 32679–32685. [Google Scholar] [CrossRef]
- Hong, S.K.; Yoon, S.; Moelling, C.; Arthan, D.; Park, J.I. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J. Biol. Chem. 2009, 284, 33006–33018. [Google Scholar] [CrossRef]
- Wu, P.K.; Hong, S.K.; Veeranki, S.; Karkhanis, M.; Starenki, D.; Plaza, J.A.; Park, J.I. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol. Cell. Biol. 2013, 33, 4051–4067. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.K.; Park, J.I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar]
- Karkhanis, M.; Park, J.I. Sp1 regulates Raf/MEK/ERK-induced p21(CIP1) transcription in TP53-mutated cancer cells. Cell. Signal. 2015, 27, 479–486. [Google Scholar] [CrossRef]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms shaping the role of ERK1/2 in cellular senescence (Review). Mol. Med. Rep. 2019, 19, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, A.; Lahav, G. The puzzling interplay between p53 and Sp1. Aging 2017, 9, 1355–1356. [Google Scholar] [CrossRef] [PubMed]
- Nishiki, Y.; Farb, T.B.; Friedrich, J.; Bokvist, K.; Mirmira, R.G.; Maier, B. Characterization of a novel polyclonal anti-hypusine antibody. Springerplus 2013, 2, 421. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, A.E.; Wu, P.-K.; Park, J.-I. eIF5A-Independent Role of DHPS in p21CIP1 and Cell Fate Regulation. Int. J. Mol. Sci. 2021, 22, 13187. https://doi.org/10.3390/ijms222413187
Becker AE, Wu P-K, Park J-I. eIF5A-Independent Role of DHPS in p21CIP1 and Cell Fate Regulation. International Journal of Molecular Sciences. 2021; 22(24):13187. https://doi.org/10.3390/ijms222413187
Chicago/Turabian StyleBecker, Andrew E., Pui-Kei Wu, and Jong-In Park. 2021. "eIF5A-Independent Role of DHPS in p21CIP1 and Cell Fate Regulation" International Journal of Molecular Sciences 22, no. 24: 13187. https://doi.org/10.3390/ijms222413187
APA StyleBecker, A. E., Wu, P.-K., & Park, J.-I. (2021). eIF5A-Independent Role of DHPS in p21CIP1 and Cell Fate Regulation. International Journal of Molecular Sciences, 22(24), 13187. https://doi.org/10.3390/ijms222413187