
The Link between Gut Dysbiosis Caused by a High-Fat Diet and Hearing Loss




{kind=link}
{kind=link}
Abstract
1. Introduction
2. Gut Microbiota
2.1. Gut Microbiota and Inflammation
2.2. Lipopolysaccharides
2.3. Short-Chain Fatty Acids
3. How Can Diet Affect the Intestinal Barrier?
High-Fat Diet and Intestinal Permeability
4. Interactions between the Gut and Distant Organs
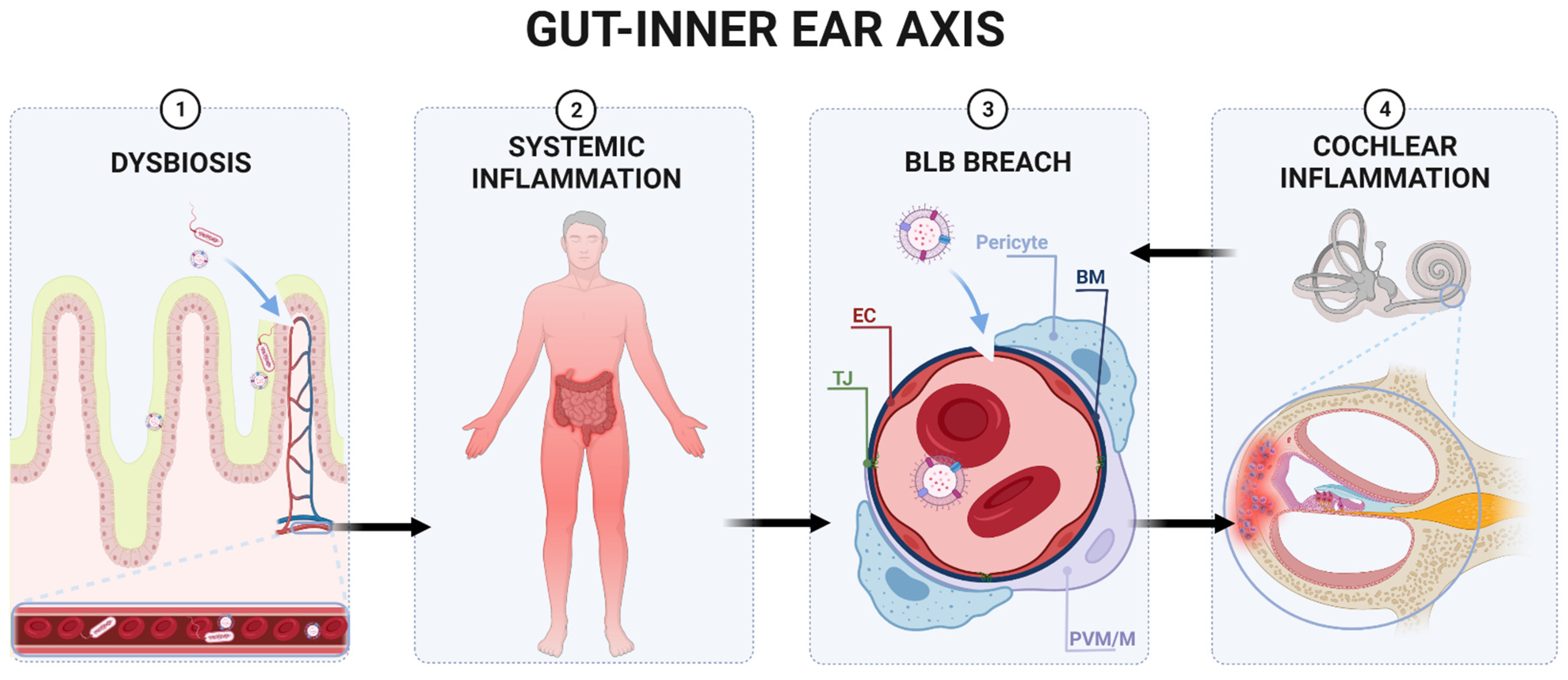
5. Could There Be a Similar Connection to the Inner Ear?
5.1. The Blood–Labyrinth Barrier
5.2. Cochlear Inflammation
5.3. Innate Cochlear Immunity and BLB
6. The Gut–Inner Ear Axis: Clinical and Experimental Evidence
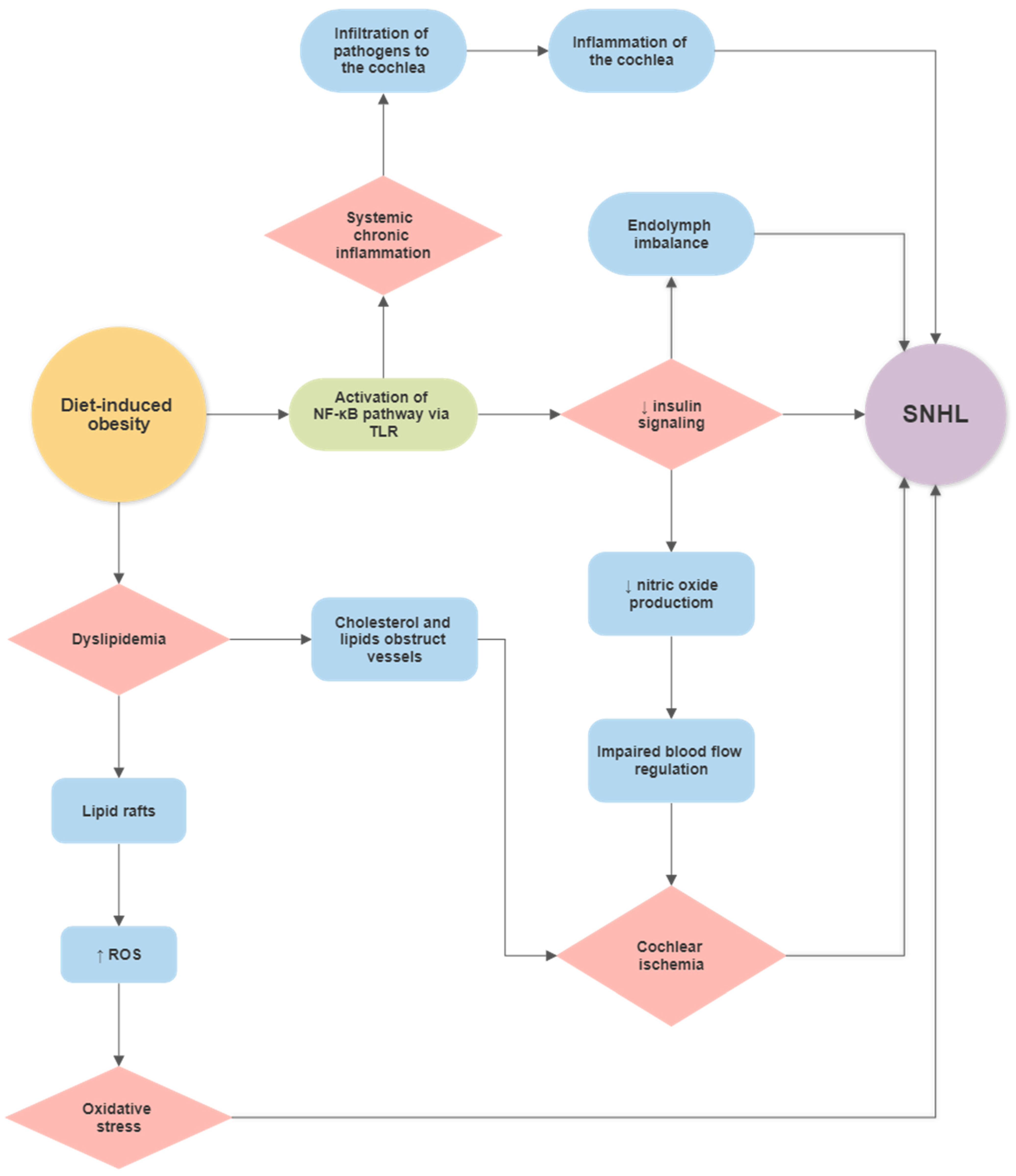
Diet-Induced Obesity (DIO) and Hearing Loss
7. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Lin, F.R.; Metter, E.J.; O’Brien, R.J.; Resnick, S.M.; Zonderman, A.B.; Ferrucci, L. Hearing loss and incident dementia. Arch. Neurol. 2011, 68, 214–220. [Google Scholar] [CrossRef]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef]
- World Health Organization. World Report on Hearing. Available online: https://www.who.int/publications/i/item/world-report-on-hearing (accessed on 2 July 2021).
- World Health Organization. Global Costs of Unadressed Hearing Loss and Cost-Effectiveness of Interventions-Executive Summary; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Croll, P.H.; Voortman, T.; Vernooij, M.W.; Baatenburg de Jong, R.J.; Lin, F.R.; Rivadeneira, F.; Ikram, M.A.; Goedegebure, A. The association between obesity, diet quality and hearing loss in older adults. Aging 2019, 11, 48–62. [Google Scholar] [CrossRef]
- Dhanda, N.; Taheri, S. A narrative review of obesity and hearing loss. Int. J. Obes. 2017, 41, 1066–1073. [Google Scholar] [CrossRef]
- Hu, H.; Tomita, K.; Kuwahara, K.; Yamamoto, M.; Uehara, A.; Kochi, T.; Eguchi, M.; Okazaki, H.; Hori, A.; Sasaki, N.; et al. Obesity and risk of hearing loss: A prospective cohort study. Clin. Nutr. 2020, 39, 870–875. [Google Scholar] [CrossRef]
- Hwang, J.H.; Hsu, C.J.; Yu, W.H.; Liu, T.C.; Yang, W.S. Diet-induced obesity exacerbates auditory degeneration via hypoxia, inflammation, and apoptosis signaling pathways in CD/1 mice. PLoS ONE 2013, 8, e60730. [Google Scholar] [CrossRef]
- Kim, S.H.; Won, Y.S.; Kim, M.G.; Baek, Y.J.; Oh, I.H.; Yeo, S.G. Relationship between obesity and hearing loss. Acta Oto-Laryngol. 2016, 136, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Scinicariello, F.; Carroll, Y.; Eichwald, J.; Decker, J.; Breysse, P.N. Association of Obesity with Hearing Impairment in Adolescents. Sci. Rep. 2019, 9, 1877. [Google Scholar] [CrossRef]
- Yang, J.R.; Hidayat, K.; Chen, C.L.; Li, Y.H.; Xu, J.Y.; Qin, L.Q. Body mass index, waist circumference, and risk of hearing loss: A meta-analysis and systematic review of observational study. Environ. Health Prev. Med. 2020, 25, 25. [Google Scholar] [CrossRef]
- Evans, M.B.; Tonini, R.; Shope, C.D.; Oghalai, J.S.; Jerger, J.F.; Insull, W., Jr.; Brownell, W.E. Dyslipidemia and auditory function. Otol. Neurotol. 2006, 27, 609–614. [Google Scholar] [CrossRef]
- Gopinath, B.; Flood, V.M.; Teber, E.; McMahon, C.M.; Mitchell, P. Dietary intake of cholesterol is positively associated and use of cholesterol-lowering medication is negatively associated with prevalent age-related hearing loss. J. Nutr. 2011, 141, 1355–1361. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Choo, O.S.; Kim, Y.J.; Gil, E.S.; Jang, J.H.; Kang, Y.; Choung, Y.H. Atorvastatin prevents hearing impairment in the presence of hyperlipidemia. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118850. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, G.; Namyslowski, G.; Morawski, K.; Strojek, K. Cochlear dysfunction and diabetic microangiopathy. Scand. Audiol. Suppl. 2001, 30, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C. The Association of Insulin Resistance and Metabolic Syndrome with Age-Related Hearing Loss. J. Diabetes Metab. 2014, 5, 2. [Google Scholar] [CrossRef]
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef]
- Tang, T.H.; Hwang, J.H.; Yang, T.H.; Hsu, C.J.; Wu, C.C.; Liu, T.C. Can Nutritional Intervention for Obesity and Comorbidities Slow Down Age-Related Hearing Impairment? Nutrients 2019, 11, 1668. [Google Scholar] [CrossRef]
- Bibbo, S.; Ianiro, G.; Giorgio, V.; Scaldaferri, F.; Masucci, L.; Gasbarrini, A.; Cammarota, G. The role of diet on gut microbiota composition. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4742–4749. [Google Scholar]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- Davis, C.D. The Gut Microbiome and Its Role in Obesity. Nutr. Today 2016, 51, 167–174. [Google Scholar] [CrossRef]
- Hakansson, A.; Molin, G. Gut microbiota and inflammation. Nutrients 2011, 3, 637–682. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Slyepchenko, A.; Maes, M.; Machado-Vieira, R.; Anderson, G.; Solmi, M.; Sanz, Y.; Berk, M.; Kohler, C.A.; Carvalho, A.F. Intestinal Dysbiosis, Gut Hyperpermeability and Bacterial Translocation: Missing Links Between Depression, Obesity and Type 2 Diabetes. Curr. Pharm. Des. 2016, 22, 6087–6106. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Schloss, P.D.; Iverson, K.D.; Petrosino, J.F.; Schloss, S.J. The dynamics of a family’s gut microbiota reveal variations on a theme. Microbiome 2014, 2, 25. [Google Scholar] [CrossRef]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70 (Suppl. S1), S38–S44. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietila, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef]
- Chelakkot, C.; Choi, Y.; Kim, D.K.; Park, H.T.; Ghim, J.; Kwon, Y.; Jeon, J.; Kim, M.S.; Jee, Y.K.; Gho, Y.S.; et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp. Mol. Med. 2018, 50, e450. [Google Scholar] [CrossRef]
- Macchione, I.G.; Lopetuso, L.R.; Ianiro, G.; Napoli, M.; Gibiino, G.; Rizzatti, G.; Petito, V.; Gasbarrini, A.; Scaldaferri, F. Akkermansia muciniphila: Key player in metabolic and gastrointestinal disorders. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8075–8083. [Google Scholar] [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; Francois, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.L.; Onnerfalt, J.; Xu, J.; Molin, G.; Ahrne, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef]
- Salguero, M.V.; Al-Obaide, M.A.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef]
- d’Hennezel, E.; Abubucker, S.; Murphy, L.O.; Cullen, T.W. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems 2017, 2, e00046-17. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Caesar, R.; Reigstad, C.S.; Backhed, H.K.; Reinhardt, C.; Ketonen, M.; Lunden, G.O.; Cani, P.D.; Backhed, F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 2012, 61, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Neff, L.M.; Suarez-Farinas, M.; Holt, P.R. Diet-induced weight loss reduces colorectal inflammation: Implications for colorectal carcinogenesis. Am. J. Clin. Nutr. 2011, 93, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.J.; van der Velden, S.; Rios-Morales, M.; van Faassen, M.J.R.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Inca, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Hitchon, C.A.; Walld, R.; Bolton, J.M.; Sareen, J.; Walker, J.R.; Graff, L.A.; Patten, S.B.; Singer, A.; Lix, L.M.; et al. Increased Burden of Psychiatric Disorders in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 360–368. [Google Scholar] [CrossRef]
- Timm, S.; Svanes, C.; Janson, C.; Sigsgaard, T.; Johannessen, A.; Gislason, T.; Jogi, R.; Omenaas, E.; Forsberg, B.; Torén, K. Place of upbringing in early childhood as related to inflammatory bowel diseases in adulthood: A population-based cohort study in Northern Europe. Eur. J. Epidemiol. 2014, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Simonsen, J.; Nielsen, N.M.; Frisch, M. Cesarean section and offspring’s risk of inflammatory bowel disease: A national cohort study. Inflamm. Bowel Dis. 2012, 18, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.R.; Russell, R.K.; Wilson, M.L.; Gilmour, W.H.; Satsangi, J.; Wilson, D.C. Systematic review: The role of breastfeeding in the development of pediatric inflammatory bowel disease. J. Pediatr. 2009, 155, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Lysy, J.; Hoshen, M.; Avitan, M.; Goldin, E.; Israeli, E. Childhood hygiene is associated with the risk for inflammatory bowel disease: A population-based study. Off. J. Am. Coll. Gastroenterol. ACG 2008, 103, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Kronman, M.P.; Zaoutis, T.E.; Haynes, K.; Feng, R.; Coffin, S.E. Antibiotic exposure and IBD development among children: A population-based cohort study. Pediatrics 2012, 130, e794–e803. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.Y.; Blanchard, J.F.; Bernstein, C.N. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Off. J. Am. Coll. Gastroenterol. ACG 2010, 105, 2687–2692. [Google Scholar] [CrossRef]
- Virta, L.; Auvinen, A.; Helenius, H.; Huovinen, P.; Kolho, K.-L. Association of repeated exposure to antibiotics with the development of pediatric Crohn’s disease—a nationwide, register-based Finnish case-control study. Am. J. Epidemiol. 2012, 175, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.; Silverstein, M.; Sandborn, W.; Tremaine, W.; Harmsen, W.; Zinsmeister, A.R. Ulcerative colitis in Olmsted County, Minnesota, 1940–1993: Incidence, prevalence, and survival. Gut 2000, 46, 336–343. [Google Scholar] [CrossRef]
- Loftus Jr, E.V.; Silverstein, M.D.; Sandborn, W.J.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeister, A.R. Crohn’s disease in Olmsted County, Minnesota, 1940–1993: Incidence, prevalence, and survival. Gastroenterology 1998, 114, 1161–1168. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Farzaei, M.H.; Rahimi, R.; Abdollahi, M. The role of dietary polyphenols in the management of inflammatory bowel disease. Curr. Pharm. Biotechnol. 2015, 16, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef]
- Olendzki, B.C.; Silverstein, T.D.; Persuitte, G.M.; Ma, Y.; Baldwin, K.R.; Cave, D. An anti-inflammatory diet as treatment for inflammatory bowel disease: A case series report. Nutr. J. 2014, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.-G.; Ahlbom, A.; Hellers, G. Diet and inflammatory bowel disease: A case-control study. Epidemiology 1992, 3, 47–52. [Google Scholar] [CrossRef]
- Dlugosz, A.; Nowak, P.; D’Amato, M.; Mohammadian Kermani, G.; Nystrom, J.; Abdurahman, S.; Lindberg, G. Increased serum levels of lipopolysaccharide and antiflagellin antibodies in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2015, 27, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Tavakoli, P.; Vollmer-Conna2, U.; Hadzi-Pavlovic, D.; Grimm, M. A Review of Inflammatory Bowel Disease: A Model of Microbial, Immune and Neuropsychological Integration. Public Health Rev. 2021, 42, 1603990. [Google Scholar] [CrossRef] [PubMed]
- Duchmann, R.; Kaiser, I.; Hermann, E.; Mayet, W.; Ewe, K.; Meyer zum Büschenfelde, K.H. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 102, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.J.; O’Connor, H.J.; Pinder, I.; Hamilton, I.; Johnston, D.; Axon, A.T. Double blind controlled trial of oral vancomycin as adjunctive treatment in acute exacerbations of idiopathic colitis. Gut 1985, 26, 1380–1384. [Google Scholar] [CrossRef]
- Sartor, R.B. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: Antibiotics, probiotics, and prebiotics. Gastroenterology 2004, 126, 1620–1633. [Google Scholar] [CrossRef]
- Khan, K.J.; Ullman, T.A.; Ford, A.C.; Abreu, M.T.; Abadir, A.; Marshall, J.K.; Talley, N.J.; Moayyedi, P. Antibiotic therapy in inflammatory bowel disease: A systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Mennigen, R.; Heptner, B.; Senninger, N.; Rijcken, E. Temporary fecal diversion in the management of colorectal and perianal Crohn’s disease. Gastroenterol. Res. Pract. 2015, 2015, 286315. [Google Scholar] [CrossRef]
- Singh, S.; Ding, N.S.; Mathis, K.L.; Dulai, P.S.; Farrell, A.M.; Pemberton, J.H.; Hart, A.L.; Sandborn, W.J.; Loftus, E.V., Jr. Systematic review with meta-analysis: Faecal diversion for management of perianal Crohn’s disease. Aliment Pharm. Ther. 2015, 42, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Allan, R.N.; Keighley, M.R.B. Effect of Fecal Diversion Alone on Perianal Crohn’s Disease. World J. Surg. 2000, 24, 1258–1263. [Google Scholar] [CrossRef]
- Mayer, E.A. Gut feelings: The emerging biology of gut-brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef]
- Lyte, M.; Vulchanova, L.; Brown, D.R. Stress at the intestinal surface: Catecholamines and mucosa–bacteria interactions. Cell Tissue Res. 2011, 343, 23–32. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Tannock, G.W. Exploring the relationships between intestinal microflora and inflammatory conditions of the human bowel and spine. Antonie Leeuwenhoek 2002, 81, 529–535. [Google Scholar] [CrossRef]
- Zamani, S.; Hesam Shariati, S.; Zali, M.R.; Asadzadeh Aghdaei, H.; Sarabi Asiabar, A.; Bokaie, S.; Nomanpour, B.; Sechi, L.A.; Feizabadi, M.M. Detection of enterotoxigenic Bacteroides fragilis in patients with ulcerative colitis. Gut Pathog. 2017, 9, 53. [Google Scholar] [CrossRef]
- Becker, H.E.F.; Jamin, C.; Bervoets, L.; Boleij, A.; Xu, P.; Pierik, M.J.; Stassen, F.R.M.; Savelkoul, P.H.M.; Penders, J.; Jonkers, D. Higher Prevalence of Bacteroides fragilis in Crohn’s Disease Exacerbations and Strain-Dependent Increase of Epithelial Resistance. Front. Microbiol. 2021, 12, 598232. [Google Scholar] [CrossRef]
- Valguarnera, E.; Wardenburg, J.B. Good Gone Bad: One Toxin Away From Disease for Bacteroides fragilis. J. Mol. Biol. 2020, 432, 765–785. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.J.; Gonipeta, B.; Parvataneni, S.; Appledorn, D.M.; Patial, S.; Sharma, D.; Gangur, V.; Amalfitano, A.; Parameswaran, N. Regulation of lipopolysaccharide-induced inflammatory response and endotoxemia by beta-arrestins. J. Cell. Physiol. 2010, 225, 406–416. [Google Scholar] [CrossRef]
- Janssen, A.W.; Kersten, S. Potential mediators linking gut bacteria to metabolic health: A critical view. J. Physiol. 2017, 595, 477–487. [Google Scholar] [CrossRef]
- Greiner, T.; Backhed, F. Effects of the gut microbiota on obesity and glucose homeostasis. Trends Endocrinol. Metab. 2011, 22, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol. 2007, 8, 1. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Fogelman, A.M. Lipopolysaccharide-induced inhibition of scavenger receptor expression in human monocyte-macrophages is mediated through tumor necrosis factor-α. J. Immunol. 1992, 148, 112–116. [Google Scholar] [PubMed]
- Wang, T.; Qin, L.; Liu, B.; Liu, Y.; Wilson, B.; Eling, T.E.; Langenbach, R.; Taniura, S.; Hong, J.S. Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J. Neurochem. 2004, 88, 939–947. [Google Scholar] [CrossRef]
- Henderson, D.; Bielefeld, E.C.; Harris, K.C.; Hu, B.H. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006, 27, 1–19. [Google Scholar] [CrossRef]
- Wong, A.C.; Ryan, A.F. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 2015, 7, 58. [Google Scholar] [CrossRef]
- Banfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed]
- Mohri, H.; Ninoyu, Y.; Sakaguchi, H.; Hirano, S.; Saito, N.; Ueyama, T. Nox3-Derived Superoxide in Cochleae Induces Sensorineural Hearing Loss. J. Neurosci. 2021, 41, 4716–4731. [Google Scholar] [CrossRef]
- Benkafadar, N.; Francois, F.; Affortit, C.; Casas, F.; Ceccato, J.C.; Menardo, J.; Venail, F.; Malfroy-Camine, B.; Puel, J.L.; Wang, J. ROS-Induced Activation of DNA Damage Responses Drives Senescence-Like State in Postmitotic Cochlear Cells: Implication for Hearing Preservation. Mol. Neurobiol. 2019, 56, 5950–5969. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; McGillicuddy, F.C.; Anderson, P.D.; Hinkle, C.C.; Shah, R.; Pruscino, L.; Tabita-Martinez, J.; Sellers, K.F.; Rickels, M.R.; Reilly, M.P. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes 2010, 59, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lin, S.; Zheng, B.; Cheung, P.C.K. Short-chain fatty acids in control of energy metabolism. Crit. Rev. Food Sci. Nutr. 2018, 58, 1243–1249. [Google Scholar] [CrossRef]
- Kim, K.N.; Yao, Y.; Ju, S.Y. Short Chain Fatty Acids and Fecal Microbiota Abundance in Humans with Obesity: A Systematic Review and Meta-Analysis. Nutrients 2019, 11, 2512. [Google Scholar] [CrossRef]
- Hartstra, A.V.; Bouter, K.E.; Bäckhed, F.; Nieuwdorp, M. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care 2015, 38, 159–165. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Bach Knudsen, K.E.; Laerke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef]
- Klampfer, L.; Huang, J.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Inhibition of Interferon γ Signaling by the Short Chain Fatty Acid Butyrate11Montefiore Medical Center New Research Initiative Award to LK and the American Cancer Society Institutional Research Grant to LK (ACS IRG# 98-274-01), UO1 CA88104 (to LA), and P30-13330 from NCI. Mol. Cancer Res. 2003, 1, 855–862. [Google Scholar]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, P.; Langholz, E.; Hollander, D.; Thornberg, K.; Orholm, M.; Katz, K.D.; Binder, V. Intestinal permeability in patients with Crohn’s disease and ulcerative colitis and their first degree relatives. Gut 1994, 35, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Kabi, A.; Nickerson, K.P.; McDonald, C. Combinatorial effects of diet and genetics on inflammatory bowel disease pathogenesis. Inflamm. Bowel Dis. 2015, 21, 912–922. [Google Scholar] [CrossRef]
- Paik, J.; Fierce, Y.; Treuting, P.M.; Brabb, T.; Maggio-Price, L. High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a-/- male mice. J. Nutr. 2013, 143, 1240–1247. [Google Scholar] [CrossRef]
- Caradonna, L.; Amati, L.; Magrone, T.; Pellegrino, N.M.; Jirillo, E.; Caccavo, D. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: Biological and clinical significance. J. Endotoxin Res. 2000, 6, 205–214. [Google Scholar]
- Argollo, M.; Gilardi, D.; Peyrin-Biroulet, C.; Chabot, J.F.; Peyrin-Biroulet, L.; Danese, S. Comorbidities in inflammatory bowel disease: A call for action. Lancet Gastroenterol. Hepatol. 2019, 4, 643–654. [Google Scholar] [CrossRef]
- Karmody, C.S.; Valdez, T.A.; Desai, U.; Blevins, N.H. Sensorineural hearing loss in patients with inflammatory bowel disease. Am. J. Otolaryngol. 2009, 30, 166–170. [Google Scholar] [CrossRef]
- Solmaz, F.; Unal, F.; Apuhan, T. Celiac disease and sensorineural hearing loss in children. Acta Oto-Laryngol. 2012, 132, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hizli, S.; Karabulut, H.; Ozdemir, O.; Acar, B.; Abaci, A.; Dagli, M.; Karasen, R.M. Sensorineural hearing loss in pediatric celiac patients. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 65–68. [Google Scholar] [CrossRef]
- Poritz, L.S.; Garver, K.I.; Green, C.; Fitzpatrick, L.; Ruggiero, F.; Koltun, W.A. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J. Surg. Res. 2007, 140, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Feng, W.; Wang, Y.; Liu, Y.; Barker, D.F.; Barve, S.S.; McClain, C.J. The type of dietary fat modulates intestinal tight junction integrity, gut permeability, and hepatic toll-like receptor expression in a mouse model of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2012, 36, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Stenman, L.K.; Holma, R.; Korpela, R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J. Gastroenterol. 2012, 18, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Lenicek, M.; Duricova, D.; Komarek, V.; Gabrysova, B.; Lukas, M.; Smerhovsky, Z.; Vitek, L. Bile acid malabsorption in inflammatory bowel disease: Assessment by serum markers. Inflamm. Bowel Dis. 2011, 17, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Ye, D.; Dokladny, K.; Ma, T.Y. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J. Immunol. 2008, 180, 5653–5661. [Google Scholar] [CrossRef]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef]
- Cao, S.; Zhang, Q.; Wang, C.; Wu, H.; Jiao, L.; Hong, Q.; Hu, C. LPS challenge increased intestinal permeability, disrupted mitochondrial function and triggered mitophagy of piglets. Innate Immun. 2018, 24, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; von der Weid, P.Y. Lipopolysaccharides modulate intestinal epithelial permeability and inflammation in a species-specific manner. Gut Microbes 2020, 11, 421–432. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Mani, V.; Hollis, J.H.; Gabler, N.K. Dietary oil composition differentially modulates intestinal endotoxin transport and postprandial endotoxemia. Nutr. Metab. 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Laugerette, F.; Vors, C.; Geloen, A.; Chauvin, M.A.; Soulage, C.; Lambert-Porcheron, S.; Peretti, N.; Alligier, M.; Burcelin, R.; Laville, M.; et al. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 2011, 22, 53–59. [Google Scholar] [CrossRef]
- Kawano, Y.; Nakae, J.; Watanabe, N.; Kikuchi, T.; Tateya, S.; Tamori, Y.; Kaneko, M.; Abe, T.; Onodera, M.; Itoh, H. Colonic Pro-inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab. 2016, 24, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Ye, D.; Said, H.M.; Ma, T.Y. IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am. J. Pathol. 2010, 177, 2310–2322. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.G.; Leonel, A.J.; Aguilar, E.C.; Batista, N.V.; Alves, A.C.; Coimbra, C.C.; Ferreira, A.V.; de Faria, A.M.; Cara, D.C.; Alvarez Leite, J.I. The combination of high-fat diet-induced obesity and chronic ulcerative colitis reciprocally exacerbates adipose tissue and colon inflammation. Lipids Health Dis. 2011, 10, 204. [Google Scholar] [CrossRef]
- Yang, R.; Han, X.; Uchiyama, T.; Watkins, S.K.; Yaguchi, A.; Delude, R.L.; Fink, M.P. IL-6 is essential for development of gut barrier dysfunction after hemorrhagic shock and resuscitation in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G621–G629. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef]
- Zou, J.; Chassaing, B.; Singh, V.; Pellizzon, M.; Ricci, M.; Fythe, M.D.; Kumar, M.V.; Gewirtz, A.T. Fiber-Mediated Nourishment of Gut Microbiota Protects against Diet-Induced Obesity by Restoring IL-22-Mediated Colonic Health. Cell Host Microbe 2018, 23, 41–53.e44. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Vyas, D.; Clark, A.T.; Woolsey, C.A.; Clark, J.A.; Hotchkiss, R.S.; Buchman, T.G.; Coopersmith, C.M. Intestine-specific overexpression of IL-10 improves survival in polymicrobial sepsis. Shock 2008, 29, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Loren, V.; Cabre, E.; Ojanguren, I.; Domenech, E.; Pedrosa, E.; Garcia-Jaraquemada, A.; Manosa, M.; Manye, J. Interleukin-10 Enhances the Intestinal Epithelial Barrier in the Presence of Corticosteroids through p38 MAPK Activity in Caco-2 Monolayers: A Possible Mechanism for Steroid Responsiveness in Ulcerative Colitis. PLoS ONE 2015, 10, e0130921. [Google Scholar] [CrossRef]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.M.; Diepolder, H.; Marquardt, A.; Jagla, W.; Popp, A.; et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G827–G838. [Google Scholar] [CrossRef]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, T.M.; Myklebust, R.; Whybrow, A.; Jenkins, R. Epithelial integrity, cell death and cell loss in mammalian small intestine. Histol. Histopathol. 1999, 14, 257–267. [Google Scholar] [CrossRef]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef]
- Qin, X.; Caputo, F.J.; Xu, D.Z.; Deitch, E.A. Hydrophobicity of mucosal surface and its relationship to gut barrier function. Shock 2008, 29, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, S.M.; Qin, X.; Lu, Q.; Feketeova, E.; Palange, D.C.; Dong, W.; Sheth, S.U.; Lee, M.A.; Reino, D.; Xu, D.Z.; et al. Loss of the intestinal mucus layer in the normal rat causes gut injury but not toxic mesenteric lymph nor lung injury. Shock 2010, 34, 475–481. [Google Scholar] [CrossRef]
- Liu, J.J.; Wong, K.; Thiesen, A.L.; Mah, S.J.; Dieleman, L.A.; Claggett, B.; Saltzman, J.R.; Fedorak, R.N. Increased epithelial gaps in the small intestines of patients with inflammatory bowel disease: Density matters. Gastrointest. Endosc. 2011, 73, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, R.; Duckworth, C.A.; Moussata, D.; Gloeckner, A.; Lim, L.G.; Goetz, M.; Pritchard, D.M.; Galle, P.R.; Neurath, M.F.; Watson, A.J. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut 2012, 61, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [PubMed]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.B.; Ducroc, R.; Regnault, B.; Kun Tan, C.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Cleary, J.L.; Condren, A.R.; Zink, K.E.; Sanchez, L.M. Calling all hosts: Bacterial communication in situ. Chem 2017, 2, 334–358. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Gut-brain axis in 2016: Brain-gut-microbiota axis-mood, metabolism and behaviour. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 69–70. [Google Scholar] [CrossRef]
- Karst, S.M. The influence of commensal bacteria on infection with enteric viruses. Nat. Rev. Microbiol. 2016, 14, 197–204. [Google Scholar] [CrossRef]
- Turroni, S.; Brigidi, P.; Cavalli, A.; Candela, M. Microbiota-Host Transgenomic Metabolism, Bioactive Molecules from the Inside. J. Med. Chem. 2018, 61, 47–61. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Fasano, A.; Shea-Donohue, T. Mechanisms of disease: The role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 416–422. [Google Scholar] [CrossRef]
- Pott, J.; Hornef, M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 2012, 13, 684–698. [Google Scholar] [CrossRef]
- Cani, P.D.; Knauf, C. How gut microbes talk to organs: The role of endocrine and nervous routes. Mol. Metab. 2016, 5, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Vaishnava, S.; Hooper, L.V. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 2009, 31, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Behrendt, C.L.; Hooper, L.V. Innate immune responses to commensal bacteria in the gut epithelium. J. Pediatr. Gastroenterol. Nutr. 2008, 46 (Suppl. S1), E10–E11. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Microbial recognition by Toll-like receptors. J. Dermatol. Sci. 2004, 34, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Coquenlorge, S.; Duchalais, E.; Chevalier, J.; Cossais, F.; Rolli-Derkinderen, M.; Neunlist, M. Modulation of lipopolysaccharide-induced neuronal response by activation of the enteric nervous system. J. Neuroinflamm. 2014, 11, 202. [Google Scholar] [CrossRef]
- Yamamoto, M.; Guo, D.H.; Hernandez, C.M.; Stranahan, A.M. Endothelial Adora2a Activation Promotes Blood-Brain Barrier Breakdown and Cognitive Impairment in Mice with Diet-Induced Insulin Resistance. J. Neurosci. 2019, 39, 4179–4192. [Google Scholar] [CrossRef]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef]
- Hsuchou, H.; Kastin, A.J.; Mishra, P.K.; Pan, W. C-reactive protein increases BBB permeability: Implications for obesity and neuroinflammation. Cell. Physiol. Biochem. 2012, 30, 1109–1119. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Salbaum, J.M.; Luo, M.; Blanchard, E.t.; Taylor, C.M.; Welsh, D.A.; Berthoud, H.R. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol. Psychiatry 2015, 77, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Grenham, S.; Clarke, G.; Cryan, J.F.; Dinan, T.G. Brain-gut-microbe communication in health and disease. Front. Physiol. 2011, 2, 94. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Gareau, M.G.; Wine, E.; Rodrigues, D.M.; Cho, J.H.; Whary, M.T.; Philpott, D.J.; Macqueen, G.; Sherman, P.M. Bacterial infection causes stress-induced memory dysfunction in mice. Gut 2011, 60, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Serino, M.; Blasco, G.; Puig, J.; Daunis-i-Estadella, J.; Ricart, W.; Burcelin, R.; Fernandez-Aranda, F.; Portero-Otin, M. Gut Microbiota Interacts With Brain Microstructure and Function. J. Clin. Endocrinol. Metab. 2015, 100, 4505–4513. [Google Scholar] [CrossRef]
- Malan-Muller, S.; Valles-Colomer, M.; Raes, J.; Lowry, C.A.; Seedat, S.; Hemmings, S.M.J. The Gut Microbiome and Mental Health: Implications for Anxiety- and Trauma-Related Disorders. Omics J. Integr. Biol. 2018, 22, 90–107. [Google Scholar] [CrossRef]
- Peterson, C.T. Dysfunction of the Microbiota-Gut-Brain Axis in Neurodegenerative Disease: The Promise of Therapeutic Modulation With Prebiotics, Medicinal Herbs, Probiotics, and Synbiotics. J. Evid.-Based Integr. Med. 2020, 25, 2515690X20957225. [Google Scholar] [CrossRef] [PubMed]
- Cerovic, M.; Forloni, G.; Balducci, C. Neuroinflammation and the Gut Microbiota: Possible Alternative Therapeutic Targets to Counteract Alzheimer’s Disease? Front. Aging Neurosci. 2019, 11, 284. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Wang, M.D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 2017, 61, 101–130. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease. Front. Microbiol. 2016, 7, 1544. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef]
- Salat-Foix, D.; Tran, K.; Ranawaya, R.; Meddings, J.; Suchowersky, O. Increased intestinal permeability and Parkinson disease patients: Chicken or egg? Can. J. Neurol. Sci. 2012, 39, 185–188. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, H.E.; Bai, Y.M.; Tsai, S.J.; Su, T.P.; Chen, T.J.; Wang, Y.P.; Chen, M.H. Inflammatory bowel disease is associated with higher dementia risk: A nationwide longitudinal study. Gut 2021, 70, 85–91. [Google Scholar] [CrossRef]
- Chen, Q.Q.; Haikal, C.; Li, W.; Li, J.Y. Gut Inflammation in Association With Pathogenesis of Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 166. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- de Aquino, C.C.; Leitao, R.A.; Oliveira Alves, L.A.; Coelho-Santos, V.; Guerrant, R.L.; Ribeiro, C.F.; Malva, J.O.; Silva, A.P.; Oria, R.B. Effect of Hypoproteic and High-Fat Diets on Hippocampal Blood-Brain Barrier Permeability and Oxidative Stress. Front. Nutr. 2018, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Hajiluian, G.; Nameni, G.; Shahabi, P.; Mesgari-Abbasi, M.; Sadigh-Eteghad, S.; Farhangi, M.A. Vitamin D administration, cognitive function, BBB permeability and neuroinflammatory factors in high-fat diet-induced obese rats. Int. J. Obes. 2017, 41, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.; Kundu, P.; Rooney, W.D.; Raber, J. The Effect of High Fat Diet on Cerebrovascular Health and Pathology: A Species Comparative Review. Molecules 2021, 26, 3406. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, J.; Rao, Y.; Chen, J.; Chen, K.; Tang, Y. Lipopolysaccharide disrupts the cochlear blood-labyrinth barrier by activating perivascular resident macrophages and up-regulating MMP-9. Int. J. Pediatr. Otorhinolaryngol. 2019, 127, 109656. [Google Scholar] [CrossRef]
- Hirose, K.; Hartsock, J.J.; Johnson, S.; Santi, P.; Salt, A.N. Systemic lipopolysaccharide compromises the blood-labyrinth barrier and increases entry of serum fluorescein into the perilymph. J. Assoc. Res. Otolaryngol. JARO 2014, 15, 707–719. [Google Scholar] [CrossRef]
- Shi, X. Pathophysiology of the cochlear intrastrial fluid-blood barrier (review). Hear. Res. 2016, 338, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, S.; Abbott, N.J.; Shi, X.; Steyger, P.S.; Dabdoub, A. Delivery of therapeutics to the inner ear: The challenge of the blood-labyrinth barrier. Sci. Transl. Med. 2019, 11, eaao0935. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina-Glushkovskaya, O.; Esmat, A.; Bragin, D.; Bragina, O.; Shirokov, A.A.; Navolokin, N.; Yang, Y.; Abdurashitov, A.; Khorovodov, A.; Terskov, A.; et al. Phenomenon of music-induced opening of the blood-brain barrier in healthy mice. Proc. Biol. Sci. 2020, 287, 20202337. [Google Scholar] [CrossRef] [PubMed]
- Shi, X. Physiopathology of the cochlear microcirculation. Hear. Res. 2011, 282, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Neng, L.; Zhang, F.; Kachelmeier, A.; Shi, X. Endothelial cell, pericyte, and perivascular resident macrophage-type melanocyte interactions regulate cochlear intrastrial fluid-blood barrier permeability. J. Assoc. Res. Otolaryngol. JARO 2013, 14, 175–185. [Google Scholar] [CrossRef]
- Shi, X. Resident macrophages in the cochlear blood-labyrinth barrier and their renewal via migration of bone-marrow-derived cells. Cell Tissue Res. 2010, 342, 21–30. [Google Scholar] [CrossRef]
- Lassale, C.; Vullo, P.; Cadar, D.; Batty, G.D.; Steptoe, A.; Zaninotto, P. Association of inflammatory markers with hearing impairment: The English Longitudinal Study of Ageing. Brain Behav. Immun. 2020, 83, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.B.; Zuo, J. The Contribution of Immune Infiltrates to Ototoxicity and Cochlear Hair Cell Loss. Front. Cell. Neurosci. 2017, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- So, H.; Kim, H.; Lee, J.H.; Park, C.; Kim, Y.; Kim, E.; Kim, J.K.; Yun, K.J.; Lee, K.M.; Lee, H.Y.; et al. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J. Assoc. Res. Otolaryngol. JARO 2007, 8, 338–355. [Google Scholar] [CrossRef]
- Adams, J.C.; Seed, B.; Lu, N.; Landry, A.; Xavier, R.J. Selective activation of nuclear factor kappa B in the cochlea by sensory and inflammatory stress. Neuroscience 2009, 160, 530–539. [Google Scholar] [CrossRef]
- Hu, B.h.; Zhang, C. Immune System and Macrophage Activation in the Cochlea: Implication for Therapeutic Intervention. In New Therapies to Prevent or Cure Auditory Disorders; Pucheu, S., Radziwon, K.E., Salvi, R., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 113–134. [Google Scholar]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Hu, B.H.; Zhang, C.; Frye, M.D. Immune cells and non-immune cells with immune function in mammalian cochleae. Hear. Res. 2018, 362, 14–24. [Google Scholar] [CrossRef]
- Wu, Y.X.; Zhu, G.X.; Liu, X.Q.; Sun, F.; Zhou, K.; Wang, S.; Wang, C.M.; Jia, J.W.; Song, J.T.; Lu, L.J. Noise alters guinea pig’s blood-labyrinth barrier ultrastructure and permeability along with a decrease of cochlear Claudin-5 and Occludin. BMC Neurosci. 2014, 15, 136. [Google Scholar] [CrossRef]
- Chan, J.; Hirose, K. Permeability of the Blood-Labyrinth Barrier in Mice Following Acoustic Injury. Otolaryngol.—Head Neck Surg. 2016, 131, P104–P105. [Google Scholar] [CrossRef]
- Neng, L.; Zhang, J.; Yang, J.; Zhang, F.; Lopez, I.A.; Dong, M.; Shi, X. Structural changes in thestrial blood-labyrinth barrier of aged C57BL/6 mice. Cell Tissue Res. 2015, 361, 685–696. [Google Scholar] [CrossRef]
- Yang, C.H.; Hwang, C.F.; Chuang, J.H.; Lian, W.S.; Wang, F.S.; Yang, M.Y. Systemic toll-like receptor 9 agonist CpG oligodeoxynucleotides exacerbates aminoglycoside ototoxicity. Hear. Res. 2021, 411, 108368. [Google Scholar] [CrossRef]
- Dhukhwa, A.; Bhatta, P.; Sheth, S.; Korrapati, K.; Tieu, C.; Mamillapalli, C.; Ramkumar, V.; Mukherjea, D. Targeting Inflammatory Processes Mediated by TRPVI and TNF-alpha for Treating Noise-Induced Hearing Loss. Front. Cell. Neurosci. 2019, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Nicotera, P.; Orrenius, S. Ca2+ and cell death. Ann. N. Y. Acad. Sci. 1992, 648, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Wu, C.C.; Hsu, C.J.; Liu, T.C.; Yang, W.S. Association of central obesity with the severity and audiometric configurations of age-related hearing impairment. Obesity 2009, 17, 1796–1801. [Google Scholar] [CrossRef]
- Lalwani, A.K.; Katz, K.; Liu, Y.H.; Kim, S.; Weitzman, M. Obesity is associated with sensorineural hearing loss in adolescents. Laryngoscope 2013, 123, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Rosenhall, U.; Sundh, V. Age-related hearing loss and blood pressure. Noise Health 2006, 8, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, D.H.; Lee, H.J.; Kim, H.J.; Koo, J.W.; Choi, H.G.; Park, B.; Hong, S.K. Lipid profiles and obesity as potential risk factors of sudden sensorineural hearing loss. PLoS ONE 2015, 10, e0122496. [Google Scholar] [CrossRef] [PubMed]
- Akbayir, N.; Calis, A.B.; Alkim, C.; Sokmen, H.M.; Erdem, L.; Ozbal, A.; Bolukbas, F.; Akbayir, N. Sensorineural hearing loss in patients with inflammatory bowel disease: A subclinical extraintestinal manifestation. Dig. Dis. Sci. 2005, 50, 1938–1945. [Google Scholar] [CrossRef]
- Wengrower, D.; Koslowsky, B.; Peleg, U.; Mazuz, B.; Cohen, L.; Ben-David, A.; Gross, M.; Goldin, E.; Shaul, C. Hearing Loss in Patients with Inflammatory Bowel Disease. Dig. Dis. Sci. 2016, 61, 2027–2032. [Google Scholar] [CrossRef]
- Kang, S.H.; Jung, D.J.; Choi, E.W.; Park, J.W.; Cho, K.H.; Lee, K.Y.; Do, J.Y. Visceral Fat Area Determined Using Bioimpedance Analysis Is Associated with Hearing Loss. Int. J. Med. Sci. 2015, 12, 946–951. [Google Scholar] [CrossRef][Green Version]
- Fukushima, H.; Cureoglu, S.; Schachern, P.A.; Paparella, M.M.; Harada, T.; Oktay, M.F. Effects of type 2 diabetes mellitus on cochlear structure in humans. Arch. Otolaryngol. Head Neck Surg. 2006, 132, 934–938. [Google Scholar] [CrossRef]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. 2014, 7, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Bacha, F.; Saad, R.; Gungor, N.; Arslanian, S.A. Are obesity-related metabolic risk factors modulated by the degree of insulin resistance in adolescents? Diabetes Care 2006, 29, 1599–1604. [Google Scholar] [CrossRef]
- Reaven, G.M. Insulin resistance: The link between obesity and cardiovascular disease. Med. Clin. N. Am. 2011, 95, 875–892. [Google Scholar] [CrossRef]
- Steinberger, J.; Daniels, S.R.; American Heart Association Atherosclerosis, H.; Obesity in the Young, C.; American Heart Association Diabetes, C. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: An American Heart Association scientific statement from the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism). Circulation 2003, 107, 1448–1453. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- de La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G440–G448. [Google Scholar] [CrossRef]
- Keshk, W.A.; Ibrahim, M.A.; Shalaby, S.M.; Zalat, Z.A.; Elseady, W.S. Redox status, inflammation, necroptosis and inflammasome as indispensable contributors to high fat diet (HFD)-induced neurodegeneration; Effect of N-acetylcysteine (NAC). Arch. Biochem. Biophys. 2020, 680, 108227. [Google Scholar] [CrossRef]
- Nuzzo, D.; Amato, A.; Picone, P.; Terzo, S.; Galizzi, G.; Bonina, F.P.; Mule, F.; Di Carlo, M. A Natural Dietary Supplement with a Combination of Nutrients Prevents Neurodegeneration Induced by a High Fat Diet in Mice. Nutrients 2018, 10, 1130. [Google Scholar] [CrossRef]
- Sardone, R.; Lampignano, L.; Guerra, V.; Zupo, R.; Donghia, R.; Castellana, F.; Battista, P.; Bortone, I.; Procino, F.; Castellana, M. Relationship between inflammatory food consumption and age-related hearing loss in a prospective observational cohort: Results from the salus in apulia study. Nutrients 2020, 12, 426. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Yang, Y.; Hu, Y.; Sun, Y.; Zhang, S.; Peng, W.; Zhong, Y.; Huang, X.; Kong, W. A long-term high-fat diet increases oxidative stress, mitochondrial damage and apoptosis in the inner ear of D-galactose-induced aging rats. Hear. Res. 2012, 287, 15–24. [Google Scholar] [CrossRef]
- Palbrink, A.K.; Kopietz, F.; Moren, B.; In ’t Zandt, R.; Kalinec, F.; Stenkula, K.; Goransson, O.; Holm, C.; Magnusson, M.; Degerman, E. Inner ear is a target for insulin signaling and insulin resistance: Evidence from mice and auditory HEI-OC1 cells. BMJ Open Diabetes Res. Care 2020, 8, e000820. [Google Scholar] [CrossRef]
- Wang, H.; Wang, A.X.; Aylor, K.; Barrett, E.J. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes 2013, 62, 4030–4042. [Google Scholar] [CrossRef]
- Wu, G.; Meininger, C.J. Nitric oxide and vascular insulin resistance. Biofactors 2009, 35, 21–27. [Google Scholar] [CrossRef]
- Shi, X.; Nuttall, A.L. The demonstration of nitric oxide in cochlear blood vessels in vivo and in vitro: The role of endothelial nitric oxide in venular permeability. Hear. Res. 2002, 172, 73–80. [Google Scholar] [CrossRef]
- Si, J.Q.; Zhao, H.; Yang, Y.; Jiang, Z.G.; Nuttall, A.L. Nitric oxide induces hyperpolarization by opening ATP-sensitive K(+) channels in guinea pig spiral modiolar artery. Hear. Res. 2002, 171, 167–176. [Google Scholar] [CrossRef]
- Han, Q.; Yeung, S.C.; Ip, M.S.M.; Mak, J.C.W. Dysregulation of cardiac lipid parameters in high-fat high-cholesterol diet-induced rat model. Lipids Health Dis. 2018, 17, 255. [Google Scholar] [CrossRef] [PubMed]
- Amiya, E. Interaction of hyperlipidemia and reactive oxygen species: Insights from the lipid-raft platform. World J. Cardiol. 2016, 8, 689–694. [Google Scholar] [CrossRef]
- Feron, O.; Dessy, C.; Moniotte, S.; Desager, J.P.; Balligand, J.L. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J. Clin. Investig. 1999, 103, 897–905. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kociszewska, D.; Chan, J.; Thorne, P.R.; Vlajkovic, S.M. The Link between Gut Dysbiosis Caused by a High-Fat Diet and Hearing Loss. Int. J. Mol. Sci. 2021, 22, 13177. https://doi.org/10.3390/ijms222413177
Kociszewska D, Chan J, Thorne PR, Vlajkovic SM. The Link between Gut Dysbiosis Caused by a High-Fat Diet and Hearing Loss. International Journal of Molecular Sciences. 2021; 22(24):13177. https://doi.org/10.3390/ijms222413177
Chicago/Turabian StyleKociszewska, Dagmara, Jeffrey Chan, Peter R. Thorne, and Srdjan M. Vlajkovic. 2021. "The Link between Gut Dysbiosis Caused by a High-Fat Diet and Hearing Loss" International Journal of Molecular Sciences 22, no. 24: 13177. https://doi.org/10.3390/ijms222413177
APA StyleKociszewska, D., Chan, J., Thorne, P. R., & Vlajkovic, S. M. (2021). The Link between Gut Dysbiosis Caused by a High-Fat Diet and Hearing Loss. International Journal of Molecular Sciences, 22(24), 13177. https://doi.org/10.3390/ijms222413177