
TLR4 Deficiency Affects the Microbiome and Reduces Intestinal Dysfunctions and Inflammation in Chronic Alcohol-Fed Mice

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
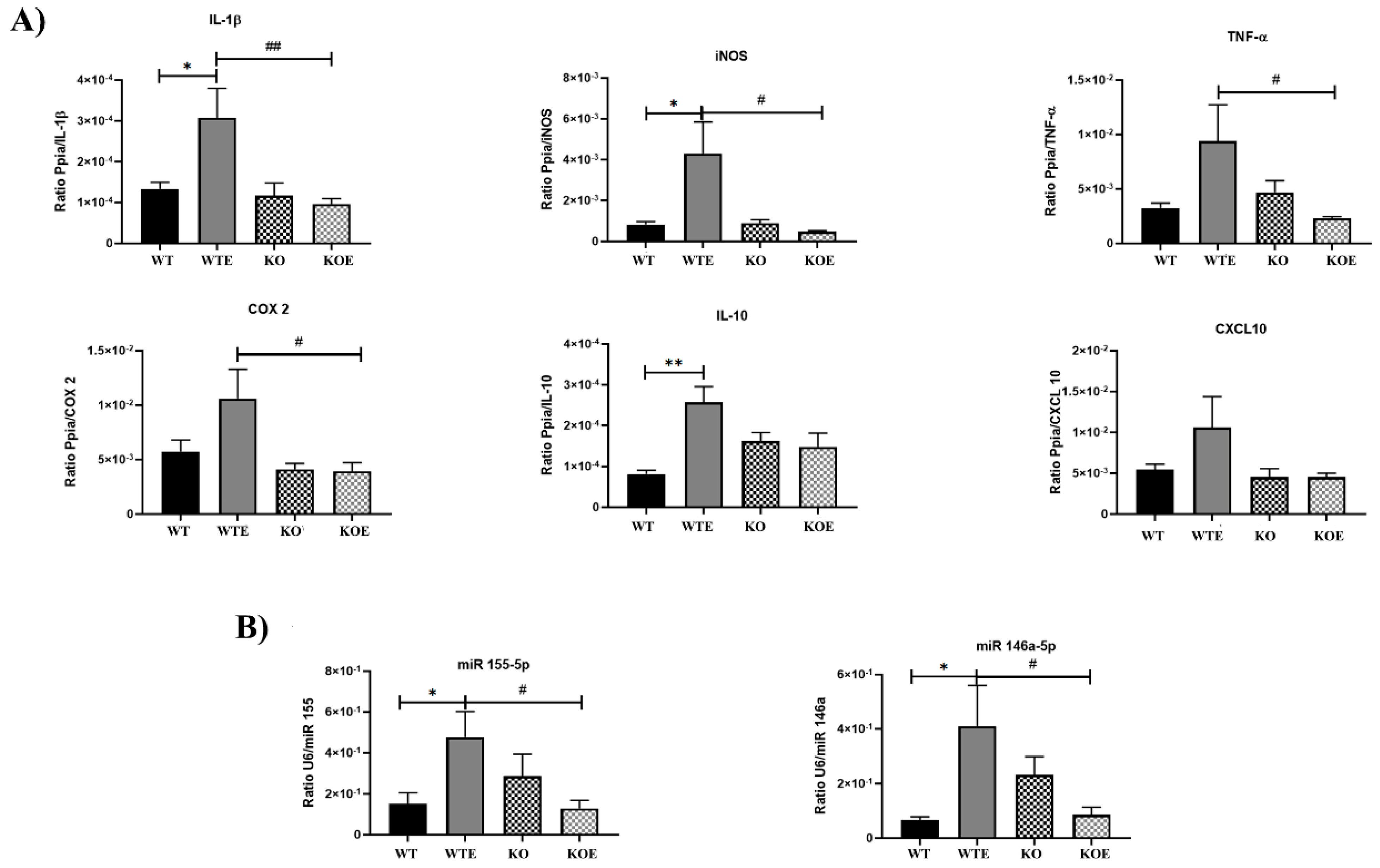
2.1. Ethanol Treatment Upregulates Inflammation in the Intestinal Genes Associated with the TLR4 Response
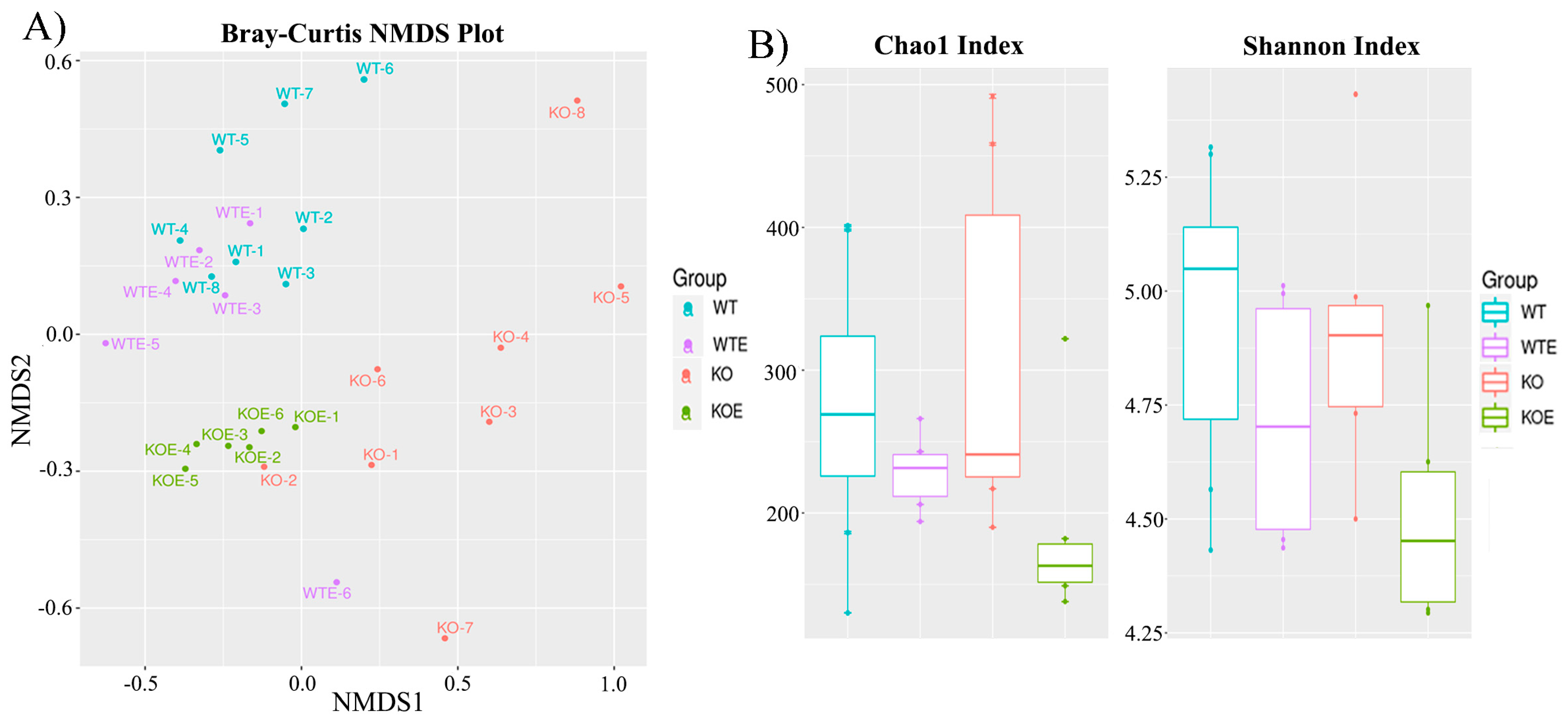
2.2. Impact of TLR4 and Ethanol Consumption on the Microbiota Community
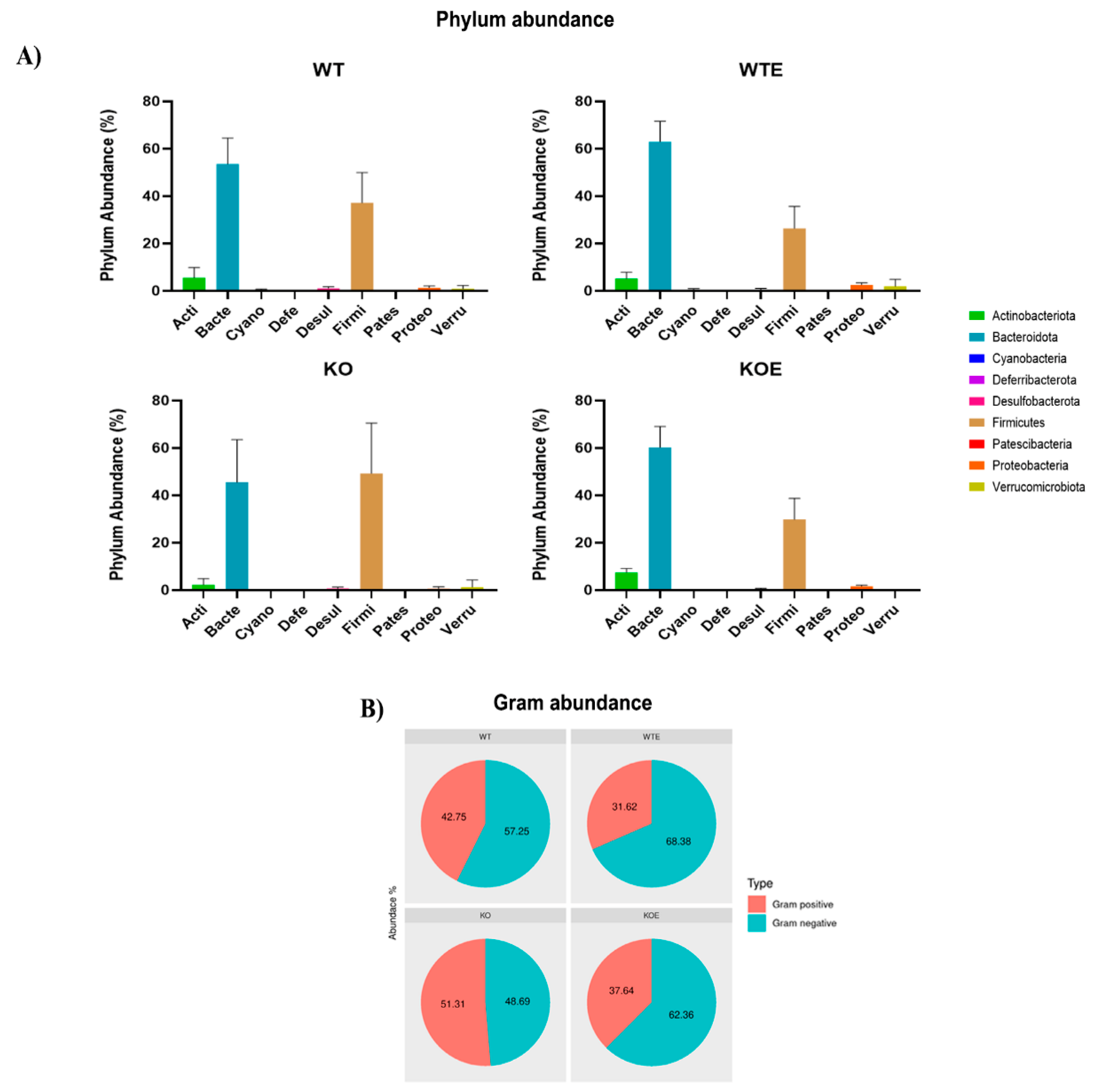
2.3. TLR4 and Chronic Alcohol Treatment Transform the Gut Microbial in Adult Male Mice
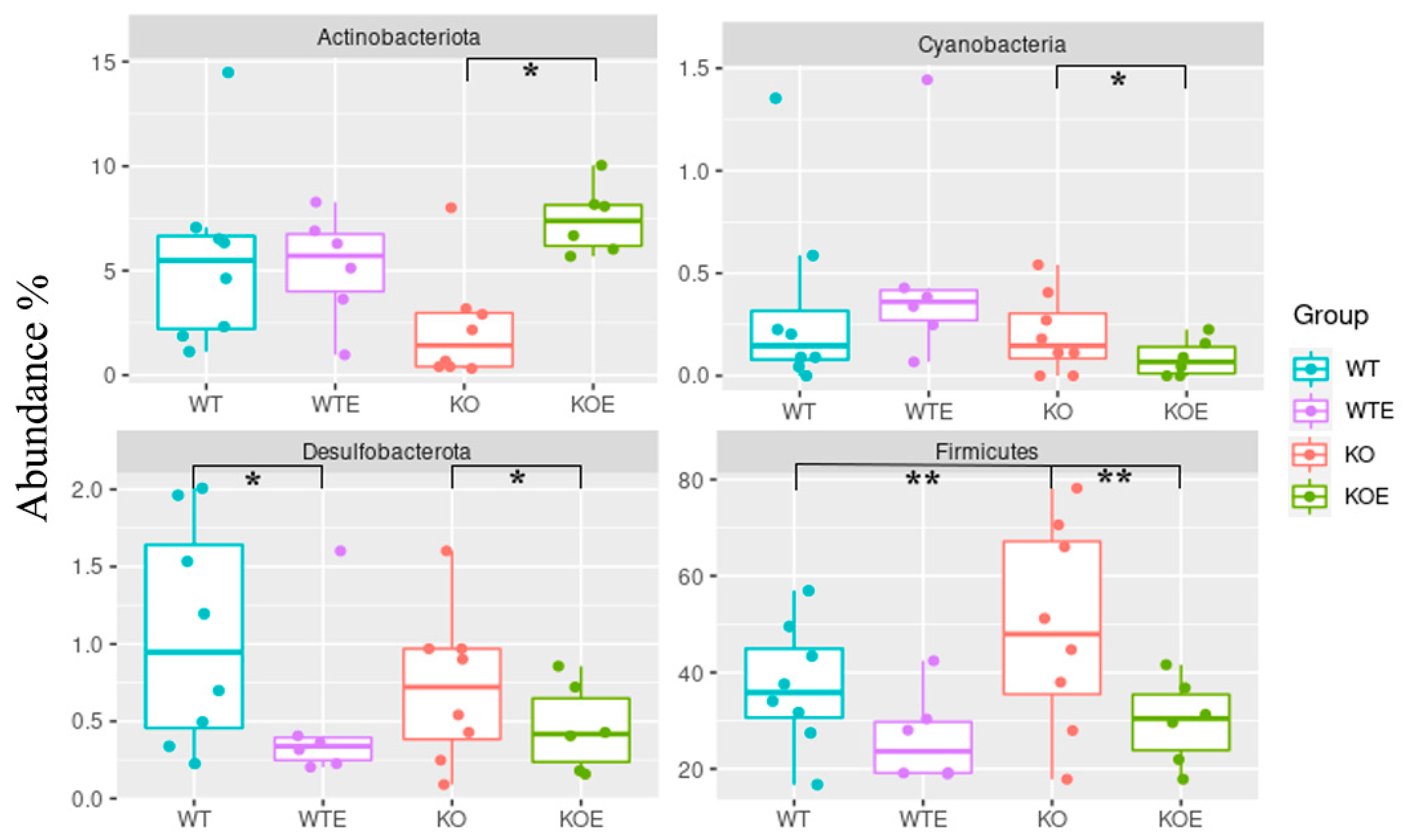
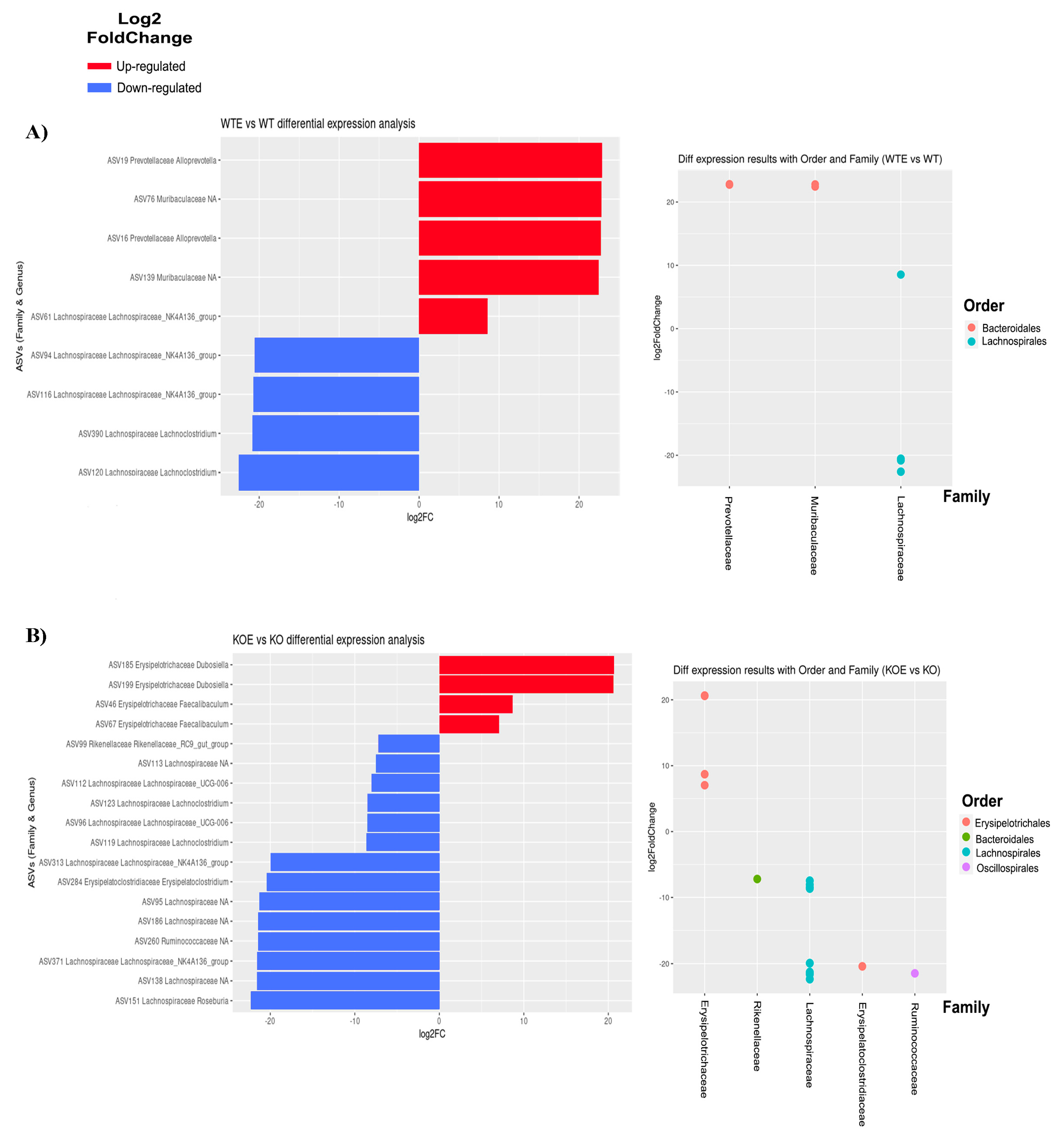
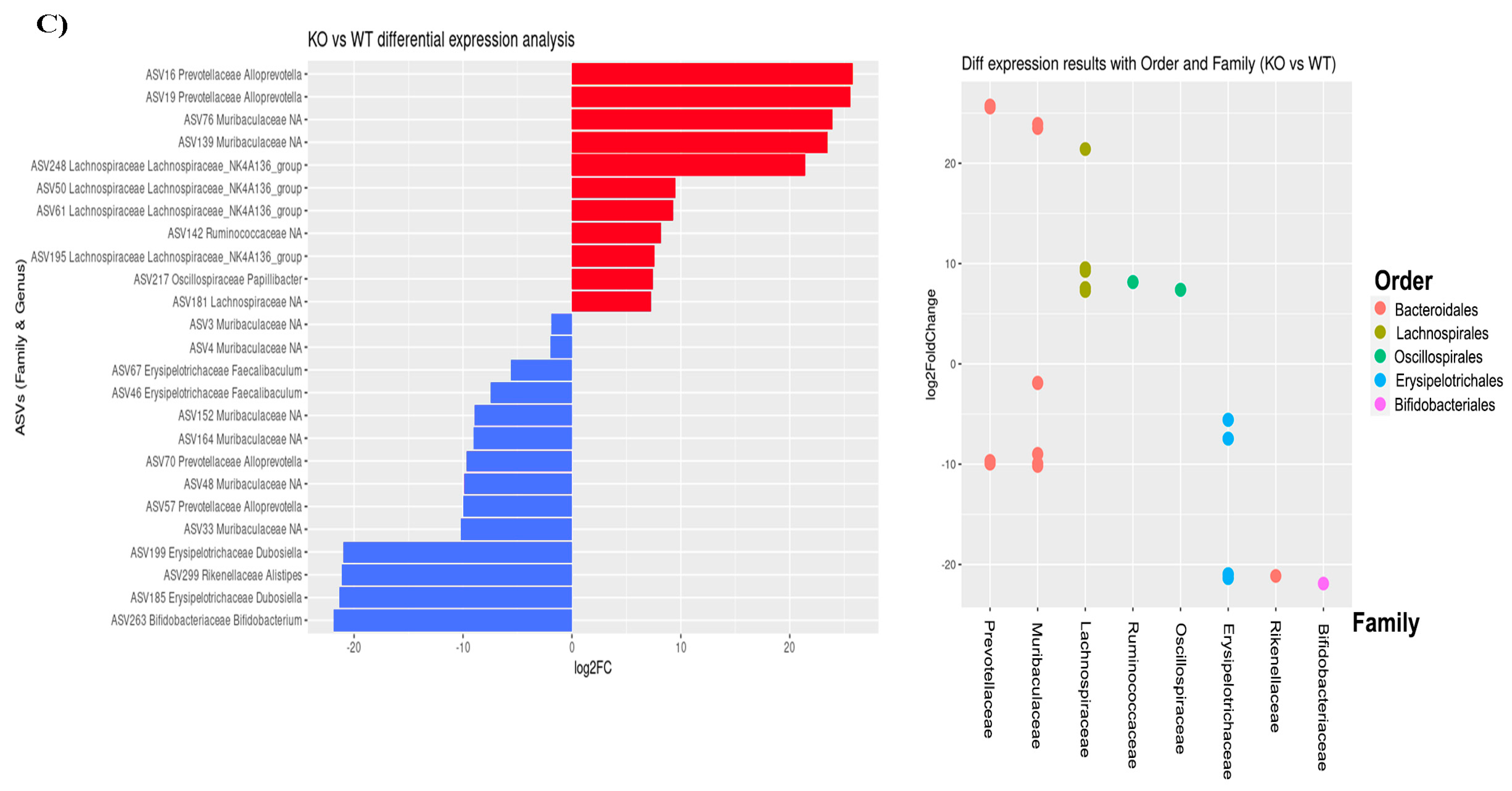
2.4. Distinct Taxa Are Affected by Ethanol and the TLR4 Response in Feces Communities
2.5. Role of TLR4 in Ethanol-Induced Alterations in Gut Structural Genes
2.6. Interactions between Inflammation and Integrity Genes and Gut Microbes
3. Discussion
4. Materials and Methods
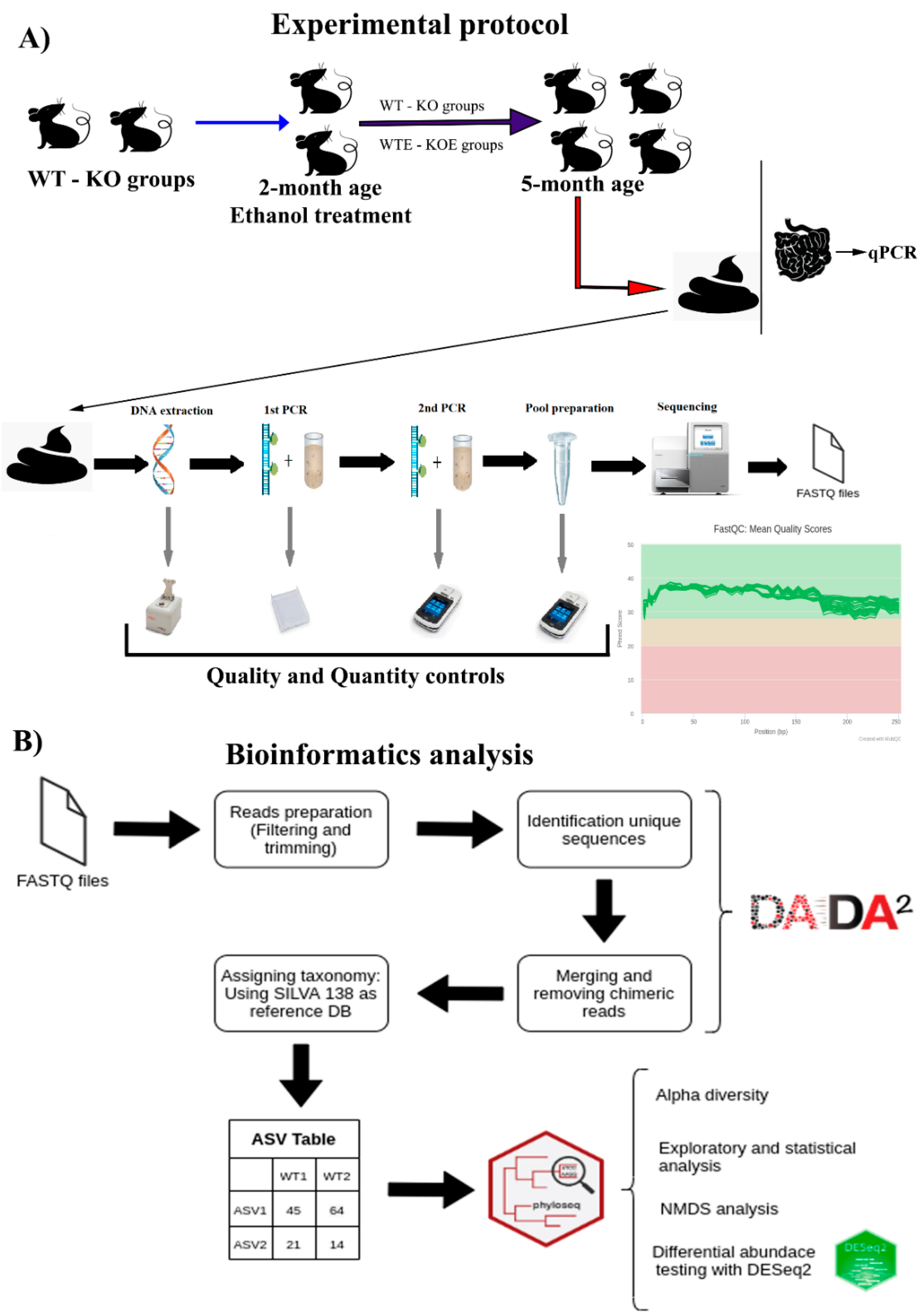
4.1. Animal Model
4.2. Alcohol Treatment
4.3. Sample Collection
4.4. Bacterial 16S rRNA Library Construction and Sequencing
4.5. 16S rRNA Gene Sequencing Analysis
4.6. Total RNA Isolation, Quantity, and Quality Determinations
4.7. Reverse Transcription and Real-Time Quantitative PCR for Total RNA and miRNA
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Wang, H.-X.; Wang, Y.-P. Gut microbiota-brain axis. Chin. Med. J. (Engl.) 2016, 129, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-Y.; Ning, M.-X.; Chen, D.-K.; Ma, W.-T. Interactions Between the Gut Microbiota and the Host Innate Immune Response Against Pathogens. Front. Immunol. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Tiffany, C.R.; Bäumler, A.J. Dysbiosis: From fiction to function. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G602–G608. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012 134 2012, 13, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wang, T.; Jin, F. Alzheimer’s disease and gut microbiota. Sci. China. Life Sci. 2016, 59, 1006–1023. [Google Scholar] [CrossRef] [Green Version]
- Srikantha, P.; Mohajeri, M.H. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 2115. [Google Scholar] [CrossRef] [Green Version]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal Microbiome: Alcohol Effects on the Composition of Intestinal Microbiota. Alcohol Res. 2015, 37, 223. [Google Scholar]
- Bode, C.; Bode, J.C. Effect of alcohol consumption on the gut. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 575–592. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Arnold, E.T.; Thomas, L.S.; Gonsky, R.; Zhou, Y.; Hu, B.; Arditi, M. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J. Biol. Chem. 2002, 277, 20431–20437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatczak, B.; Cohen, I.R. Gut feelings of safety: Tolerance to the microbiota mediated by innate immune receptors. Microbiol. Immunol. 2015, 59, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the Intricate Interaction among Toll-Like Receptors, Microbiota, and Intestinal Immunity Can Influence Gastrointestinal Pathology. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Cani, P.D.; Neyrinck, A.M.; Stärkel, P.; Jamar, F.; Mikolajczak, M.; Delzenne, N.M.; Timary, P. de Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain. Behav. Immun. 2012, 26, 911–918. [Google Scholar] [CrossRef]
- Blanco, A.M.; Vallés, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [Green Version]
- Cario, E.; Podolsky, D.K. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 2000, 68, 7010–7017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.; Delbro, D.S.; Jennische, E.; Mattsby-Baltzer, I. The role of the Lps gene in experimental ulcerative colitis in mice. APMIS 1996, 104, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Behara, R.; Swanson, G.R.; Forsyth, C.B.; Voigt, R.M.; Keshavarzian, A. Alcohol and the Intestine. Biomolecules 2015, 5, 2573–2588. [Google Scholar] [CrossRef] [PubMed]
- Ureña-Peralta, J.R.; Pérez-Moraga, R.; García-García, F.; Guerri, C. Lack of TLR4 modifies the miRNAs profile and attenuates inflammatory signaling pathways. PLoS ONE 2020, 15, e0237066. [Google Scholar] [CrossRef]
- Ureña-Peralta, J.R.; Alfonso-Loeches, S.; Cuesta-Diaz, C.M.; García-García, F.; Guerri, C. Deep sequencing and miRNA profiles in alcohol-induced neuroinflammation and the TLR4 response in mice cerebral cortex. Sci. Rep. 2018, 8, 15913. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascale, A.; Marchesi, N.; Marelli, C.; Coppola, A.; Luzi, L.; Govoni, S.; Giustina, A.; Gazzaruso, C. Microbiota and metabolic diseases. Endocrine 2018, 61, 357–371. [Google Scholar] [CrossRef]
- Velloso, L.A.; Folli, F.; Saad, M.J. TLR4 at the Crossroads of Nutrients, Gut Microbiota, and Metabolic Inflammation. Endocr. Rev. 2015, 36, 245–271. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bishehsari, F.; Magno, E.; Swanson, G.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and Gut-Derived Inflammation. Alcohol Res. 2017, 38, 163. [Google Scholar] [PubMed]
- Burgueño, J.F.; Abreu, M.T. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 263–278. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Malaguarnera, G.; Giordano, M.; Nunnari, G.; Bertino, G.; Malaguarnera, M. Gut microbiota in alcoholic liver disease: Pathogenetic role and therapeutic perspectives. World J. Gastroenterol. 2014, 20, 16639–16648. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J. Endotoxins: Lipopolysaccharides of gram-negative bacteria. Subcell. Biochem. 2010, 53, 3–25. [Google Scholar] [CrossRef]
- Im, E.; Riegler, F.M.; Pothoulakis, C.; Rhee, S.H. Elevated lipopolysaccharide in the colon evokes intestinal inflammation, aggravated in immune modulator-impaired mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G490–G497. [Google Scholar] [CrossRef] [Green Version]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Jin, Y.; Chen, G.; Ma, X.; Zhang, L. Gut Microbiota Dysbiosis Drives the Development of Colorectal Cancer. Digestion 2021, 102, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, Q.; Guo, L.; Zeng, H.; Ding, C.; Zhang, W.; Xu, D.; Wang, X.; Qiu, J.; Dong, Q.; et al. Gut Microbiota and Relevant Metabolites Analysis in Alcohol Dependent Mice. Front. Microbiol. 2018, 9, 1874. [Google Scholar] [CrossRef]
- Kriss, M.; Hazleton, K.Z.; Nusbacher, N.M.; Martin, C.G.; Lozupone, C.A. Low diversity gut microbiota dysbiosis: Drivers, functional implications and recovery. Curr. Opin. Microbiol. 2018, 44, 34–40. [Google Scholar] [CrossRef]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef] [Green Version]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Tsuruya, A.; Kuwahara, A.; Saito, Y.; Yamaguchi, H.; Tsubo, T.; Suga, S.; Inai, M.; Aoki, Y.; Takahashi, S.; Tsutsumi, E.; et al. Ecophysiological consequences of alcoholism on human gut microbiota: Implications for ethanol-related pathogenesis of colon cancer. Sci. Rep. 2016, 6, 27923. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chen, Y.; Wang, G.; Yang, Y.; Song, X.; Xiong, Z.; Zhang, H.; Lai, P.; Wang, S.; Ai, L. Lactobacillus plantarum AR113 alleviates DSS-induced colitis by regulating the TLR4/MyD88/NF-κB pathway and gut microbiota composition. J. Funct. Foods 2020, 67, 103854. [Google Scholar] [CrossRef]
- Gao, R.; Tian, S.; Wang, J.; Zhu, W. Galacto-oligosaccharides improve barrier function and relieve colonic inflammation via modulating mucosa-associated microbiota composition in lipopolysaccharides-challenged piglets. J. Anim. Sci. Biotechnol. 2021, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [Green Version]
- Betrapally, N.S.; Gillevet, P.M.; Bajaj, J.S. Changes in the Intestinal Microbiome and Alcoholic and Nonalcoholic Liver Diseases: Causes or Effects? Gastroenterology 2016, 150, 1745–1755.e3. [Google Scholar] [CrossRef] [Green Version]
- Cox, L.M.; Sohn, J.; Tyrrell, K.L.; Citron, D.M.; Lawson, P.A.; Patel, N.B.; Iizumi, T.; Perez-Perez, G.I.; Goldstein, E.J.C.; Blaser, M.J. Description of two novel members of the family Erysipelotrichaceae: Ileibacterium valens gen. nov., sp. nov. and Dubosiella newyorkensis, gen. nov., sp. nov., from the murine intestine, and emendation to the description of Faecalibaculum rodentium. Int. J. Syst. Evol. Microbiol. 2017, 67, 1247–1254. [Google Scholar] [CrossRef]
- Maccioni, L.; Gao, B.; Leclercq, S.; Pirlot, B.; Horsmans, Y.; De Timary, P.; Leclercq, I.; Fouts, D.; Schnabl, B.; Stärkel, P. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes 2020, 12, 1782157. [Google Scholar] [CrossRef]
- Seo, B.; Jeon, K.; Moon, S.; Lee, K.; Kim, W.-K.; Jeong, H.; Cha, K.H.; Lim, M.Y.; Kang, W.; Kweon, M.-N.; et al. Roseburia spp. Abundance Associates with Alcohol Consumption in Humans and Its Administration Ameliorates Alcoholic Fatty Liver in Mice. Cell Host Microbe 2020, 27, 25–40.e6. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, N.; Garrido, D. Species Deletions from Microbiome Consortia Reveal Key Metabolic Interactions between Gut Microbes. mSystems 2019, 4, e00185-19. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- Bjørkhaug, S.T.; Aanes, H.; Neupane, S.P.; Bramness, J.G.; Malvik, S.; Henriksen, C.; Skar, V.; Medhus, A.W.; Valeur, J. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes 2019, 10, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Dheer, R.; Santaolalla, R.; Davies, J.M.; Lang, J.K.; Phillips, M.C.; Pastorini, C.; Vazquez-Pertejo, M.T.; Abreu, M.T. Intestinal Epithelial Toll-Like Receptor 4 Signaling Affects Epithelial Function and Colonic Microbiota and Promotes a Risk for Transmissible Colitis. Infect. Immun. 2016, 84, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Hennezel, E.; Abubucker, S.; Murphy, L.O.; Cullen, T.W. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems 2017, 2, e00046-17. [Google Scholar] [CrossRef] [Green Version]
- Abreu, M.T.; Vora, P.; Faure, E.; Thomas, L.S.; Arnold, E.T.; Arditi, M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 2001, 167, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria with Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Xiao, L.; Chen, B.; Feng, D.; Yang, T.; Li, T.; Chen, J. TLR4 May Be Involved in the Regulation of Colonic Mucosal Microbiota by Vitamin A. Front. Microbiol. 2019, 10, 268. [Google Scholar] [CrossRef]
- Faruqui, N.A.; Prium, D.H.; Mowna, S.A.; Ullah, M.A.; Araf, Y.; Sarkar, B.; Zohora, U.S.; Rahman, M.S. Gut microorganisms and neurological disease perspectives. Future Neurol. 2021, 5, 66. [Google Scholar] [CrossRef]
- Yang, X.; Gao, X.-C.; Liu, J.; Ren, H.-Y. Effect of EPEC endotoxin and bifidobacteria on intestinal barrier function through modulation of toll-like receptor 2 and toll-like receptor 4 expression in intestinal epithelial cell-18. World J. Gastroenterol. 2017, 23, 4744–4751. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; De Saeger, C.; Delzenne, N.; de Timary, P.; Stärkel, P. Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependence. Biol. Psychiatry 2014, 76, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Moncada, C.; Núñez, M.T.; Lavandero, S.; Ponnappa, B.C.; Israel, Y. Ethanol increases tumor necrosis factor-alpha receptor-1 (TNF-R1) levels in hepatic, intestinal, and cardiac cells. Alcohol 2004, 33, 9–15. [Google Scholar] [CrossRef]
- Chen, P.; Stärkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Salaspuro, M. Microbial metabolism of ethanol and acetaldehyde and clinical consequences. Addict. Biol. 1997, 2, 35–46. [Google Scholar] [CrossRef]
- Rao, R.K. Acetaldehyde-induced barrier disruption and paracellular permeability in Caco-2 cell monolayer. Methods Mol. Biol. 2008, 447, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Nusrat, A. Cytokine regulation of tight junctions. Biochim. Biophys. Acta 2009, 1788, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tong, J.; Chang, B.; Wang, B.; Zhang, D.; Wang, B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol. Med. Rep. 2014, 9, 2352–2356. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M. E-cadherin Is Important for the Maintenance of Intestinal Epithelial Homeostasis Under Basal and Inflammatory Conditions. Dig. Dis. Sci. 2015, 60, 816–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devall, M.; Jennelle, L.T.; Bryant, J.; Bien, S.; Peters, U.; Powell, S.; Casey, G. Modeling the effect of prolonged ethanol exposure on global gene expression and chromatin accessibility in normal 3D colon organoids. PLoS ONE 2020, 15, e0227116. [Google Scholar] [CrossRef]
- Stewart, D.C.; Berrie, D.; Li, J.; Liu, X.; Rickerson, C.; Mkoji, D.; Iqbal, A.; Tan, S.; Doty, A.L.; Glover, S.C.; et al. Quantitative assessment of intestinal stiffness and associations with fibrosis in human inflammatory bowel disease. PLoS ONE 2018, 13, e0200377. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Ni, Y.; Wang, Z.; Tu, W.; Ni, L.; Zhuge, F.; Zheng, A.; Hu, L.; Zhao, Y.; Zheng, L.; et al. Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut Microbes 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Manuel, C.R.; Latuga, M.S.; Ashby, C.R., Jr.; Reznik, S.E. Immune tolerance attenuates gut dysbiosis, dysregulated uterine gene expression and high-fat diet potentiated preterm birth in mice. Am. J. Obstet. Gynecol. 2019, 220, 596.e1–596.e28. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A relevant minority for the maintenance of gut homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- Pascual, M.; Baliño, P.; Aragón, C.M.G.; Guerri, C. Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: Role of TLR4 and TLR2. Neuropharmacology 2015, 89, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Montesinos, J.; Montagud-Romero, S.; Forteza, J.; Rodríguez-Arias, M.; Miñarro, J.; Guerri, C. TLR4 response mediates ethanol-induced neurodevelopment alterations in a model of fetal alcohol spectrum disorders. J. Neuroinflam. 2017, 14, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| baseMean | Log2 Fold Change | lfcSE | Stat | p-Value | p-adj | Kingdom | Phylum | |
|---|---|---|---|---|---|---|---|---|
| WTE vs. WT | ||||||||
| ASV114 | 114.434 | −1.270113 | 0.60419 | −2.1022 | 0.03554 | 0.31985 | Bacteria | Desulfobacterota |
| KOE vs. KO | ||||||||
| ASV5 | 751.696 | 1.292251 | 0.63923 | 2.0216 | 0.04322 | 0.09724 | Bacteria | Actinobacteriota |
| ASV13 | 6441.341 | −1.388448 | 0.35495 | −3.9117 | 0.00009 | 0.00082 | Bacteria | Firmicutes |
| ASV114 | 114.434 | −1.384593 | 0.60594 | −2.2850 | 0.02231 | 0.07158 | Bacteria | Desulfobacterota |
| ASV336 | 42.115 | −1.928905 | 0.85374 | −2.2594 | 0.02386 | 0.07158 | Bacteria | Cyanobacteria |
| KO vs. WT | ||||||||
| ASV13 | 6441.341 | 1.197066 | 0.32857 | 3.6432 | 0.00027 | 0.00242 | Bacteria | Firmicutes |
| Genes | Forward (5′–3′) | Reverse (5′–3′) |
| 16S rRNA V3–V4 region | TCG TCG GCA GCG TCA GAT GTG TAT AAGAGA CAG CCT ACG GGN GGC WGC AG | GTC TCG TGG GCT CGG AGA TGT GTA TAA GAGACA GGA CTA CHV GGG TAT CTA ATC C |
| snRNA U6 | GCT TCG GCA GCA CAT ATA CTA AAA T | CGC TTC ACG AAT TTG CGT GTC AT |
| Ppia | CGC GTC TCC TTC GAG CTG TTT G | TGT AAA GTC ACC ACC CTG GCA CA |
| IL-1B | CTC ATT GTG GCT GTG GAG AA | TCT AAT GGG AAC GTC ACA CA |
| iNOS | AAT CTT GGA GCG AGT TGT GG | AAT CTC TGC CTA TCC GTC TCG |
| TNF-alfa | GAA CTG GCA GAA GAG GCA CT | AGG GTC TGG GCC ATA GAA CT |
| COX 2 | CAT TGA CCA GAG CAG AGA GAT G | GGC TTC CAG TAT TGA GGA GAA C |
| IL-10 | AGG CGC TGT CAT CGA TTT CT | ATG GCC TTG TAG ACA CCT TGG |
| CXCL10 | ATG ACG GGC CAG TGA GAA TG | TCA ACA CGT GGG CAG GAT AG |
| ITNL1 | TCC AGT CAG CAA GGC AAC AGA G | CAG GTT CTC AGC CTG GAT GTC A |
| CFTR | CCA TCA GCA AGC TGA AAG CAG G | GTA GGG TTG TAA TGC CGA GAC G |
| CDH1 | GGT CAT CAG TGT GCT CAC CTC T | GCT GTT GTG CTC AAG CCT TCA C |
| Muc2 | GCT GAC GAG TGG TTG GTG AAT G | GAT GAG GTG GCA GAC AGG AGA C |
| AQP8 | AAC TTG TGG GCT CCG CTC TCT T | ACA GCA GGG TTG AAG TGT CCA C |
| miRNA | Chromosome Location | Sequence |
| hsa-miR-146a-5p | Chr.5: 160485352 to 160485450 [+] on Build GRCh38 | UGAGAACUGAAUUCCAUGGGUU |
| mmu-miR-155-5p | Chr.16: 84714140 to 84714204 [+] on Build GRCm38 | UUAAUGCUAAUUGUGAUAGGGGU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, C.M.; Pascual, M.; Pérez-Moraga, R.; Rodríguez-Navarro, I.; García-García, F.; Ureña-Peralta, J.R.; Guerri, C. TLR4 Deficiency Affects the Microbiome and Reduces Intestinal Dysfunctions and Inflammation in Chronic Alcohol-Fed Mice. Int. J. Mol. Sci. 2021, 22, 12830. https://doi.org/10.3390/ijms222312830
Cuesta CM, Pascual M, Pérez-Moraga R, Rodríguez-Navarro I, García-García F, Ureña-Peralta JR, Guerri C. TLR4 Deficiency Affects the Microbiome and Reduces Intestinal Dysfunctions and Inflammation in Chronic Alcohol-Fed Mice. International Journal of Molecular Sciences. 2021; 22(23):12830. https://doi.org/10.3390/ijms222312830
Chicago/Turabian StyleCuesta, Carlos M., María Pascual, Raúl Pérez-Moraga, Irene Rodríguez-Navarro, Francisco García-García, Juan R. Ureña-Peralta, and Consuelo Guerri. 2021. "TLR4 Deficiency Affects the Microbiome and Reduces Intestinal Dysfunctions and Inflammation in Chronic Alcohol-Fed Mice" International Journal of Molecular Sciences 22, no. 23: 12830. https://doi.org/10.3390/ijms222312830
APA StyleCuesta, C. M., Pascual, M., Pérez-Moraga, R., Rodríguez-Navarro, I., García-García, F., Ureña-Peralta, J. R., & Guerri, C. (2021). TLR4 Deficiency Affects the Microbiome and Reduces Intestinal Dysfunctions and Inflammation in Chronic Alcohol-Fed Mice. International Journal of Molecular Sciences, 22(23), 12830. https://doi.org/10.3390/ijms222312830