
Two Opposing Functions of Angiotensin-Converting Enzyme (ACE) That Links Hypertension, Dementia, and Aging


{kind=link}
{kind=link}
Abstract
1. Introduction
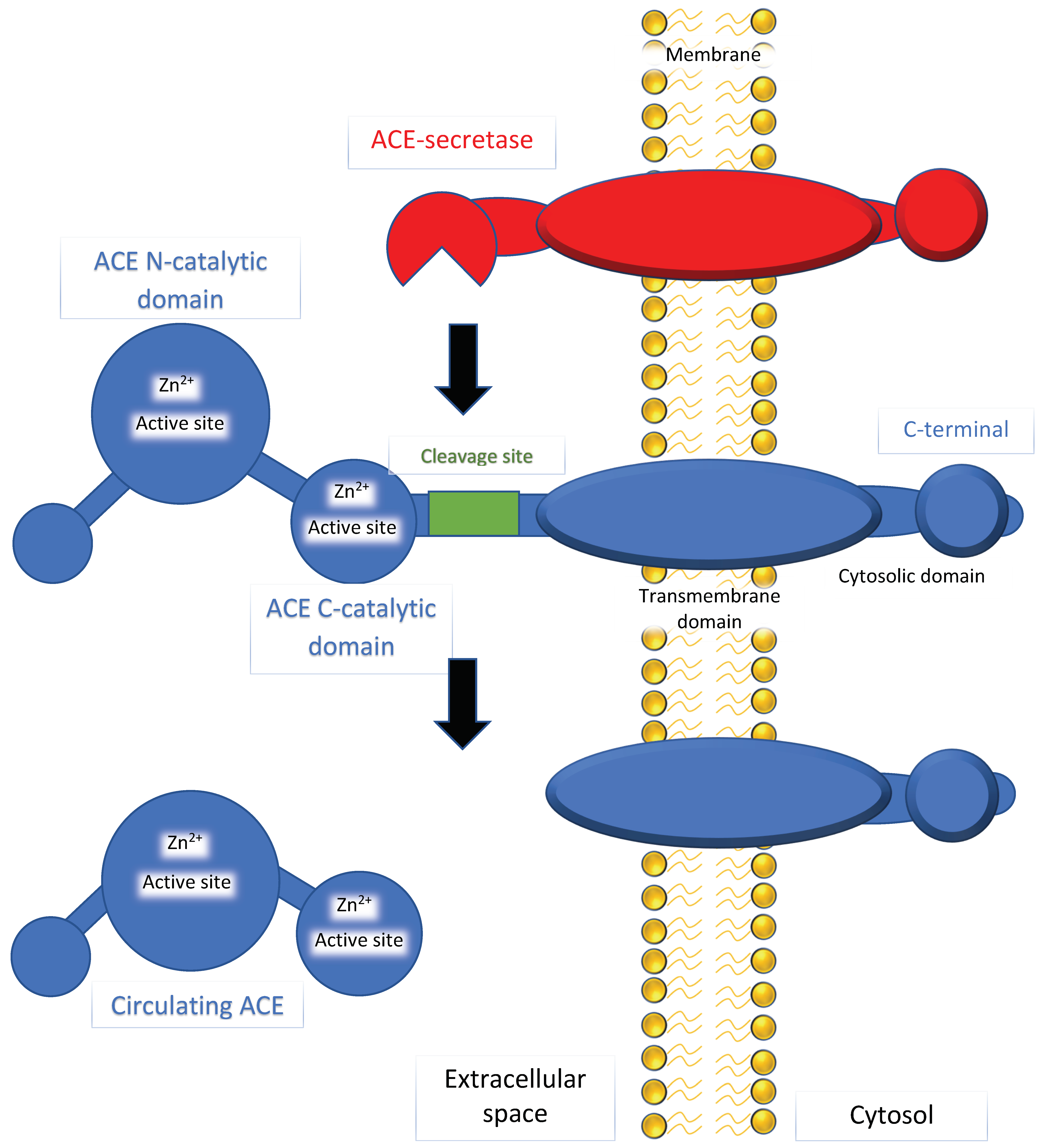
2. Mechanism of ACE
3. ACE as a Gene That Links Hypertension, AD, and Aging
3.1. ACE as a Link to Hypertension
3.2. ACE as a Link to AD and Diverse Neurological Diseases
3.3. Genetic Variations of ACE That Link to AD
3.4. Two Opposing Health Effects of ACE on AD
- (1).
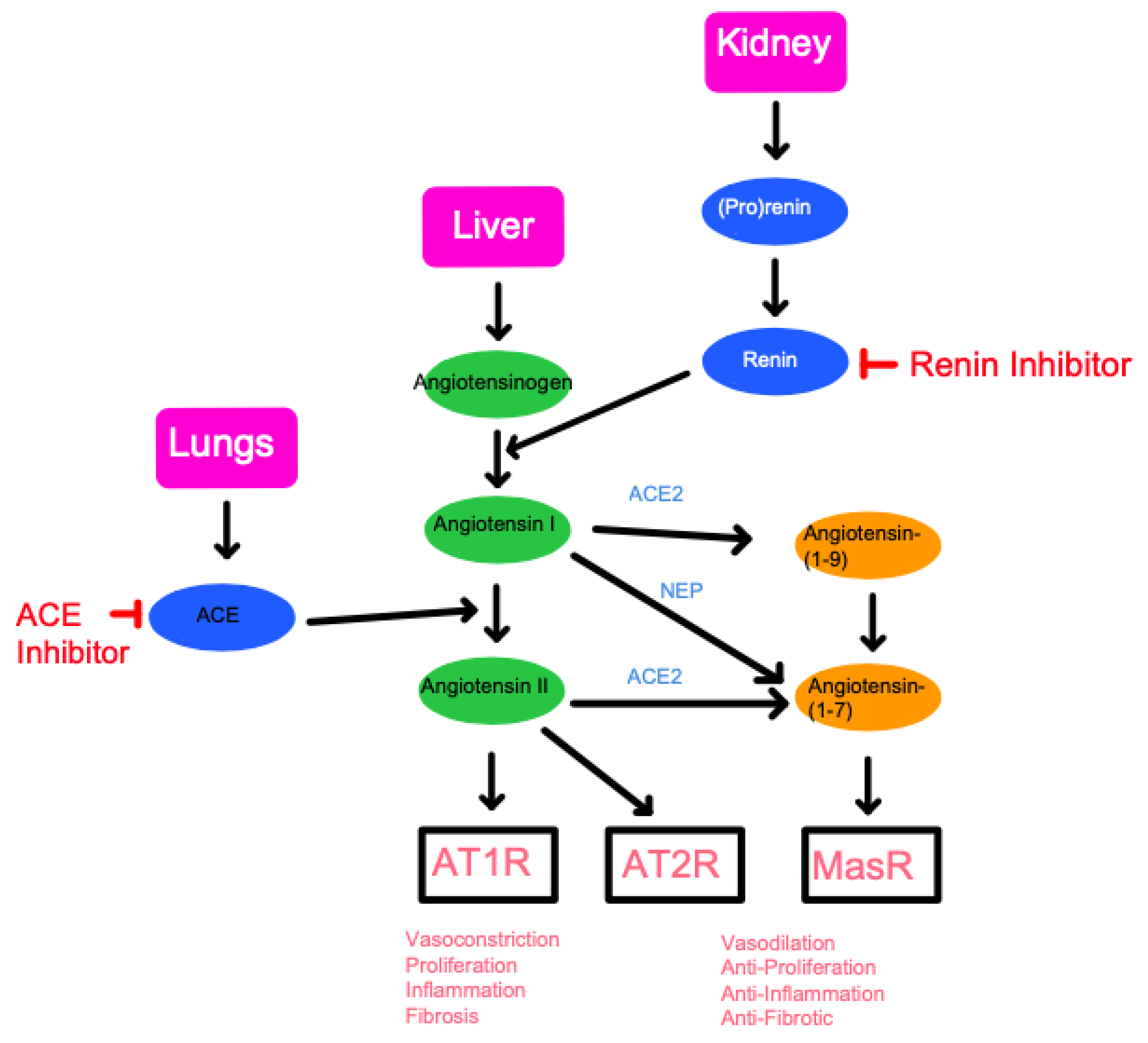
- ACE as an angiotensin-converting enzyme that makes angiotensin II in the renin–angiotensin system. The action of ACE is described in Section 2. ACE inhibitors reduce the production of angiotensin II, which results in vasodilation and reduced blood pressure. In this case, inhibiting the activities as an angiotensin-converting enzyme would relieve hypertension and hypertension-sensitive conditions, including heart failure, chronic kidney disease, or diabetes mellitus [45]. In mice, the role of the ACE/angiotensin II signaling has been investigated. A previous study suggests that a brain-penetrant ACE inhibitor, Captopril, reduces AD symptoms in the mouse AD model (Tg2576) which overexpresses a Swedish APP mutation (KM670/671NL) [46]. In another mouse AD model (5XFAD) that has five AD-linked mutations, ACE variant rs4980 (R1279Q) causes aging-dependent, Aβ-accelerated selective hippocampal neuron vulnerability and female susceptibility [43]. The Aβ-induced hippocampal neurodegeneration is rescued by the ACE inhibitor (Captopril) and AT1R inhibitor/ARB (Losartan) that can penetrate the brain [43]. The studies suggest that ACE/angiotensin II signaling causes Aβ-induced neurodegeneration and that brain-penetrant ACE inhibitor/ARB can protect the neurons. Although clinical studies remain to be completed, it is reasonable to conclude that ACE inhibitors may be beneficial to health under specific conditions.
- (2).
- ACE as an amyloid-degrading enzyme (ADE) that can hydrolyze beta-amyloid and decrease amyloid toxicity. ADE represents a group of broadly defined enzymes, currently, including 14 enzymes: ACE, acyl peptide hydrolase, aminopeptidase A, cathepsin B, endothelin-converting enzyme, glutamate carboxypeptidase II, insulin-degrading enzyme, MBP, MMP-2, MMP-9, NEP2, neprilysin, plasmin, and PreP [47,48]. ACE has an activity of beta-amyloid amyloid-degrading enzymes (ADEs) that can hydrolyze and convert Aβ1-42 to Aβ1-40 in homogenates of the mouse Tg2576 AD model and human AD autopsy [47], which is consistent with its dipeptidyl carboxypeptidase activity that cleaves the c-terminal two amino acid residues. ACE can also cleave Aβ1-40 into two smaller peptides (Aβ1-7 and Aβ8-40) [49,50,51]. In mice, inhibitions of ACE can worsen the accumulation of Aβ1-42 in the mouse model [47]. Consistently, overexpression of ACE in the brain resident microglia, peripheral myelomonocytes, and macrophages can alleviate the symptoms of the double-transgenic APPSWE/PS1ΔE9 (AD+) mice [52,53]. Thus, in the AD model system in mice, the ADE activity can reduce Aβ -induced pathologic problems. The efficacy of the ADE activity seems to be consistent with the clinical studies with increased ACE and reduced risk of AD.
3.5. ACE as a Link to Aging (Life Extension Model Systems)
3.6. ACE as a Link to Stress Resistance and the Middle-Life Crisis Theory on Aging
4. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skeggs, L.T.; Kahn, J.R.; Shumway, N.P. The preparation and function of the hypertensin-converting enzyme. J. Exp. Med. 1956, 103, 295–299. [Google Scholar] [CrossRef]
- Braun-Menendez, E.; Page, I.H. Suggested Revision of Nomenclature—Angiotensin. Science 1958, 127, 242. [Google Scholar] [CrossRef]
- Gavras, H.; Brunner, H.R.; Laragh, J.H.; Sealey, J.E.; Gavras, I.; Vukovich, R.A. An Angiotensin Converting-Enzyme Inhibitor to Identify and Treat Vasoconstrictor and Volume Factors in Hypertensive Patients. N. Engl. J. Med. 1974, 291, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Haber, E.; Barger, A.C. Experimental and clinical studies with converting enzyme inhibitor. Prog. Biochem. Pharmacol. 1976, 12, 16–32. [Google Scholar] [PubMed]
- Skrbic, R.; Igic, R. Seven decades of angiotensin (1939–2009). Peptides 2009, 30, 1945–1950. [Google Scholar] [CrossRef]
- Ehlers, M.R.; Fox, E.A.; Strydom, D.J.; Riordan, J.F. Molecular cloning of human testicular angiotensin-converting enzyme: The testis isozyme is identical to the C-terminal half of endothelial angiotensin-converting enzyme. Proc. Natl. Acad. Sci. USA 1989, 86, 7741–7745. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Musini, V.M.; Gill, R. First-line drugs for hypertension. Cochrane Database Syst. Rev. 2018, 4, CD001841. [Google Scholar] [CrossRef] [PubMed]
- Akpunonu, B.E.; Mulrow, P.J.; Hoffman, E.A. Secondary hypertension: Evaluation and treatment. Disease-a-Month 1996, 42, 609–722. [Google Scholar] [CrossRef]
- Onusko, E. Diagnosing Secondary Hypertension. Am. Fam. Physician 2003, 67, 67–74. [Google Scholar]
- Nia, B.V.; Kang, C.; Tran, M.G.; Lee, D.; Murakami, S. Meta Analysis of Human AlzGene Database: Benefits and Limitations of Using C. elegans for the Study of Alzheimer’s Disease and Co-morbid Conditions. Front. Genet. 2017, 8, 55. [Google Scholar] [CrossRef]
- Le, D.; Crouch, N.; Villanueva, A.; Phong Ta Dmitriyev, R.; Tunzi, M.; Murakami, S. Evidence-based geneti5 Editorial Board, cs and identification of key human Alzheimer’s disease alleles with co-morbidities. J. Neurol. Exp. Neurosci. 2020, 6, S1. [Google Scholar] [CrossRef]
- The Brainstorm Consortium; Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef]
- Gebre, A.K.; Altaye, B.M.; Atey, T.M.; Tuem, K.B.; Berhe, D.F. Targeting Renin–Angiotensin System Against Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Cuspidi, C.; Hering, D. Hypertension and cognitive dysfunction in elderly: Blood pressure management for this global burden. BMC Cardiovasc. Disord. 2016, 16, 208. [Google Scholar] [CrossRef]
- Antos, A.; Kwong, M.; Balmorez, T.; Villanueva, A.; Murakami, S. Unusually High Risks of COVID-19 Mortality with Age-Related Comorbidities: An Adjusted Meta-Analysis Method to Improve the Risk Assessment of Mortality Using the Comorbid Mortality Data. Infect. Dis. Rep. 2021, 13, 700–711. [Google Scholar] [CrossRef]
- Junot, C.; Théodoro, F.; Thierry, J.; Clément, G.; Wdzieczak-Bakala, J.; Ezan, E. Development of an enzyme immunoassay for a stable amidated analog of the hemoregulatory peptide acetyl-ser-asp-lys-pro. J. Immunoass. Immunochem. 2001, 22, 15–31. [Google Scholar] [CrossRef]
- Van Esch, J.H.; Tom, B.; Dive, V.; Batenburg, W.W.; Georgiadis, D.; Yiotakis, A.; van Gool, J.M.; de Bruijn, R.J.; de Vries, R.; Danser, A.J. Selective Angiotensin-Converting Enzyme C-Domain Inhibition Is Sufficient to Prevent Angiotensin I–Induced Vasoconstriction. Hypertension 2005, 45, 120–125. [Google Scholar] [CrossRef]
- Carey, R.M. Update on angiotensin AT2 receptors. Curr. Opin. Nephrol. Hypertens. 2017, 26, 91–96. [Google Scholar] [CrossRef]
- Hammer, A.; Yang, G.; Friedrich, J.; Kovacs, A.; Lee, D.-H.; Grave, K.; Jörg, S.; Alenina, N.; Grosch, J.; Winkler, J.; et al. Role of the receptor Mas in macrophage-mediated inflammation in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 14109–14114. [Google Scholar] [CrossRef] [PubMed]
- Suvila, K.; Langén, V.; Cheng, S.; Niiranen, T. Age of Hypertension Onset: Overview of Research and How to Apply in Practice. Curr. Hypertens. Rep. 2020, 22, 68. [Google Scholar] [CrossRef]
- Krause, M.W.; Fonseca, V.A.; Shah, S.V. Combination inhibition of the renin–angiotensin system: Is more better? Kidney Int. 2011, 80, 245–255. [Google Scholar] [CrossRef]
- Exner, D.V.; Dries, D.L.; Domanski, M.J.; Cohn, J.N. Lesser Response to Angiotensin-Converting–Enzyme Inhibitor Therapy in Black as Compared with White Patients with Left Ventricular Dysfunction. N. Engl. J. Med. 2001, 344, 1351–1357. [Google Scholar] [CrossRef]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Prim. 2018, 4, 18014. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Hu, J.; Miyatake, F.; Aizu, Y.; Nakagawa, H.; Nakamura, S.; Tamaoka, A.; Takahash, R.; Urakami, K.; Shoji, M. Angiotensin-converting enzyme genotype is associated with Alzheimer disease in the Japanese population. Neurosci. Lett. 1999, 277, 65–67. [Google Scholar] [CrossRef]
- Kehoe, P.G.; Russ, C.; McIlroy, S.; Williams, H.; Holmans, P.; Holmes, C.; Liolitsa, D.; Vahidassr, D.; Powell, J.; McGleenon, B.; et al. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat. Genet. 1999, 21, 71–72. [Google Scholar] [CrossRef]
- AlzGene.org. Available online: https://www.alzgene.org/geneoverview.asp?geneid=125 (accessed on 1 October 2021).
- Elkins, J.S.; Douglas, V.C.; Johnston, S.C. Alzheimer disease risk and genetic variation in ACE: A meta-analysis. Neurology 2004, 62, 363–368. [Google Scholar] [CrossRef]
- Lehmann, D.J.; Cortina-Borja, M.; Warden, D.R.; Smith, A.D.; Sleegers, K.; Prince, J.A.; Van Duijn, C.M.; Kehoe, P.G. Large Meta-Analysis Establishes the ACE Insertion-Deletion Polymorphism as a Marker of Alzheimer’s Disease. Am. J. Epidemiol. 2005, 162, 305–317. [Google Scholar] [CrossRef]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef]
- Helbecque, N.; Codron, V.; Cottel, D.; Amouyel, P. An age effect on the association of common variants of ACE with Alzheimer’s disease. Neurosci. Lett. 2009, 461, 181–184. [Google Scholar] [CrossRef]
- Gaiteri, C.; Mostafavi, S.; Honey, C.; De Jager, P.L.; Bennett, D.A. Genetic variants in Alzheimer disease—Molecular and brain network approaches. Nat. Rev. Neurol. 2016, 12, 413–427. [Google Scholar] [CrossRef]
- Corrao, S.; Coco, D.L.; Lopez, G. Cognitive impairment and stroke in elderly patients. Vasc. Health Risk Manag. 2016, 12, 105–116. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 2018, 15, 276. [Google Scholar] [CrossRef]
- Surguchov, A. Caveolin: A New Link between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef]
- Rieder, M.J.; Taylor, S.L.; Clark, A.; Nickerson, D.A. Sequence variation in the human angiotensin converting enzyme. Nat. Genet. 1999, 22, 59–62. [Google Scholar] [CrossRef]
- Kölsch, H.; Jessen, F.; Freymann, N.; Kreis, M.; Hentschel, F.; Maier, W.; Heun, R. ACE I/D polymorphism is a risk factor of Alzheimer’s disease but not of vascular dementia. Neurosci. Lett. 2005, 377, 37–39. [Google Scholar] [CrossRef]
- Sleegers, K.; Heijer, T.D.; van Dijk, E.J.; Hofman, A.; Bertoli-Avella, A.M.; Koudstaal, P.J.; Breteler, M.M.; van Duijn, C.M. ACE gene is associated with Alzheimer’s disease and atrophy of hippocampus and amygdala. Neurobiol. Aging 2005, 26, 1153–1159. [Google Scholar] [CrossRef]
- Kehoe, P.G.; Katzov, H.; Feuk, L.; Bennet, A.M.; Johansson, B.; Wiman, B.; de Faire, U.; Cairns, N.J.; Wilcock, G.K.; Brookes, A.J.; et al. Haplotypes extending across ACE are associated with Alzheimer’s disease. Hum. Mol. Genet. 2003, 12, 859–867. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Kauwe, J.S.K.; Bailey, M.H.; Ridge, P.G.; Perry, R.; Wadsworth, M.E.; Hoyt, K.L.; Staley, L.A.; Karch, C.; Harari, O.; Cruchaga, C.; et al. Genome-Wide Association Study of CSF Levels of 59 Alzheimer’s Disease Candidate Proteins: Significant Associations with Proteins Involved in Amyloid Processing and Inflammation. PLoS Genet. 2014, 10, e1004758. [Google Scholar] [CrossRef]
- Zou, K.; Yamaguchi, H.; Akatsu, H.; Sakamoto, T.; Ko, M.; Mizoguchi, K.; Gong, J.-S.; Yu, W.; Yamamoto, T.; Kosaka, K.; et al. Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition en-hances brain Abeta deposition. J. Neurosci. 2007, 27, 8628–8635. [Google Scholar] [CrossRef]
- Cuddy, L.K.; Prokopenko, D.; Cunningham, E.P.; Brimberry, R.; Song, P.; Kirchner, R.; Chapman, B.A.; Hofmann, O.; Hide, W.; Procissi, D.; et al. Aβ-accelerated neurodegeneration caused by Alzheimer’s-associated ACE variant R1279Q is rescued by angiotensin system inhibition in mice. Sci. Transl. Med. 2020, 12, eaaz2541. [Google Scholar] [CrossRef]
- Miners, S.; Ashby, E.; Baig, S.; Harrison, R.; Tayler, H.; Speedy, E.; Prince, J.A.; Love, S.; Kehoe, P.G. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl. Res. 2009, 1, 163–177. [Google Scholar]
- Mark, P.B.; Papworth, R.; Ramparsad, N.; Tomlinson, L.A.; Sawhney, S.; Black, C.; McConnachie, A.; McCowan, C. Risk factors associated with biochemically detected and hospitalised acute kidney injury in patients prescribed renin angiotensin system inhibitors. Br. J. Clin. Pharmacol. 2020, 86, 121–131. [Google Scholar] [CrossRef]
- Abdalla, S.; Langer, A.; Fu, X.; Quitterer, U. ACE Inhibition with Captopril Retards the Development of Signs of Neurodegeneration in an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 16917–16942. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Beckett, C.; Belyaev, N.D.; Turner, A.J. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 2012, 120, 167–185. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef]
- Hu, J.; Igarashi, A.; Kamata, M.; Nakagawa, H. Angiotensin-converting Enzyme Degrades Alzheimer Amyloid β-Peptide (Aβ); Retards Aβ Aggregation, Deposition, Fibril Formation; and Inhibits Cytotoxicity. J. Biol. Chem. 2001, 276, 47863–47868. [Google Scholar] [CrossRef]
- Oba, R.; Igarashi, A.; Kamata, M.; Nagata, K.; Takano, S.; Nakagawa, H. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide. Eur. J. Neurosci. 2005, 21, 733–740. [Google Scholar] [CrossRef]
- Hemming, M.L.; Selkoe, D.J. Amyloid β-Protein Is Degraded by Cellular Angiotensin-converting Enzyme (ACE) and Elevated by an ACE Inhibitor. J. Biol. Chem. 2005, 280, 37644–37650. [Google Scholar] [CrossRef]
- Bernstein, K.; Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Lopes, D.H.; Shah, K.H.; Bernstein, E.A.; Fuchs, D.-T.; Yu, J.J.-Y.; et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Investig. 2014, 124, 1000–1012. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Sheyn, J.; Hayden, E.; Li, S.; Fuchs, D.-T.; Regis, G.C.; Lopes, D.H.J.; Black, K.L.; E Bernstein, K.; Teplow, D.B.; et al. Peripherally derived angiotensin converting enzyme-enhanced macrophages alleviate Alzheimer-related disease. Brain 2020, 143, 336–358. [Google Scholar] [CrossRef]
- A Skidgel, R.; Erdös, E.G. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: A brief history, the beginning and follow-ups to early studies. Peptides 2004, 25, 521–525. [Google Scholar] [CrossRef]
- Dubreuil, P.; Fulcrand, P.; Rodriguez, M.; Laur, J.; Martinez, J. Novel activity of angiotensin-converting enzyme. Hydrolysis of cholecystokinin and gastrin analogues with release of the amidated C-terminal dipeptide. Biochem. J. 1989, 262, 125–130. [Google Scholar] [CrossRef]
- Jaspard, E.; Wei, L.; Alhenc-Gelas, F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J. Biol. Chem. 1993, 268, 9496–9503. [Google Scholar] [CrossRef]
- Kumar, S.; Dietrich, N.; Kornfeld, K. Angiotensin Converting Enzyme (ACE) Inhibitor Extends Caenorhabditis elegans Life Span. PLoS Genet. 2016, 12, e1005866. [Google Scholar] [CrossRef]
- Gabrawy, M.M.; Campbell, S.; Carbone, M.A.; Morozova, T.V.; Arya, G.H.; Turlapati, L.B.; Walston, J.D.; Starz-Gaiano, M.; Everett, L.; Mackay, T.F.C.; et al. Lisinopril Preserves Physical Resilience and Extends Life Span in a Genotype-Specific Manner in Drosophila melanogaster. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2019, 74, 1844–1852. [Google Scholar] [CrossRef]
- Spindler, S.R.; Mote, P.L.; Flegal, J.M. Combined statin and angiotensin-converting enzyme (ACE) inhibitor treatment increases the lifespan of long-lived F1 male mice. AGE 2016, 38, 379–391. [Google Scholar] [CrossRef][Green Version]
- Johnson, T.E.; Lithgow, G.J.; Murakami, S. Hypothesis: Interventions That Increase the Response to Stress Offer the Potential for Effective Life Prolongation and Increased Health. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 1996, 51, B392–B395. [Google Scholar] [CrossRef]
- Murakami, S.; E Johnson, T. A Genetic Pathway Conferring Life Extension and Resistance to UV Stress in Caenorhabditis elegans. Genetics 1996, 143, 1207–1218. [Google Scholar] [CrossRef]
- Murakami, S.; Salmon, A.; Miller, R.A. Multiplex stress resistance in cells from long-lived dwarf mice. FASEB J. 2003, 17, 1565–1576. [Google Scholar] [CrossRef]
- Murakami, S. Stress resistance in long-lived mouse models. Exp. Gerontol. 2006, 41, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Cuzzocrea, S.; Iavicoli, I.; Rizzarelli, E.; Calabrese, E.J. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Asp. Med. 2011, 32, 279–304. [Google Scholar] [CrossRef]
- Bartke, A.; Sun, L.Y.; Longo, V. Somatotropic Signaling: Trade-Offs Between Growth, Reproductive Development, and Longevity. Physiol. Rev. 2013, 93, 571–598. [Google Scholar] [CrossRef] [PubMed]
- Machino, K.; Link, C.D.; Wang, S.; Murakami, H.; Murakami, S. A semi-automated motion-tracking analysis of locomotion speed in the C. elegans transgenics overexpressing beta-amyloid in neurons. Front. Genet. 2014, 5, 202. [Google Scholar] [CrossRef]
- Dysarz, J.; Fuellen, G.; Möller, S.; Luyten, W.; Schmitz-Linneweber, C.; Saul, N. Genes implicated in Caenorhabditis elegans and human health regulate stress resistance and physical abilities in aged Caenorhabditis elegans. Biol. Lett. 2021, 17, 20200916. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Caenorhabditis elegans as a model system to study aging of learning and memory. Mol. Neurobiol. 2007, 35, 85–94. [Google Scholar] [CrossRef]
- Murakami, S.; Cabana, K.; Anderson, D. Current advances in the study of oxidative stress and age-related memory impairment in C. elegans. In Molecular Aspects of Oxidative Stress on Cell Signaling in Verte-Brates and Invertebrates; Farooqui, T., Ed.; Wiley-Blackwell Publisher: Hoboken, NJ, USA, 2011; ISBN 978-1-1180-0194-3. [Google Scholar]
- Murakami, S. Age-dependent modulation of learning and memory in C. elegans. In Handbook of Behavioral Neuroscience; Invertebrate Learning and Memory; Menzel, R., Benjamin, P.R., Eds.; Elsevier: London, UK, 2013; Chapter 12; pp. 140–150. [Google Scholar]
- Lebouvier, T.; Chen, Y.; Duriez, P.; Pasquier, F.; Bordet, R. Antihypertensive agents in Alzheimer’s disease: Beyond vascular protection. Expert Rev. Neurother. 2020, 20, 175–187. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, D.; Brown, L.; Malik, K.; Murakami, S. Two Opposing Functions of Angiotensin-Converting Enzyme (ACE) That Links Hypertension, Dementia, and Aging. Int. J. Mol. Sci. 2021, 22, 13178. https://doi.org/10.3390/ijms222413178
Le D, Brown L, Malik K, Murakami S. Two Opposing Functions of Angiotensin-Converting Enzyme (ACE) That Links Hypertension, Dementia, and Aging. International Journal of Molecular Sciences. 2021; 22(24):13178. https://doi.org/10.3390/ijms222413178
Chicago/Turabian StyleLe, Duc, Lindsay Brown, Kundan Malik, and Shin Murakami. 2021. "Two Opposing Functions of Angiotensin-Converting Enzyme (ACE) That Links Hypertension, Dementia, and Aging" International Journal of Molecular Sciences 22, no. 24: 13178. https://doi.org/10.3390/ijms222413178
APA StyleLe, D., Brown, L., Malik, K., & Murakami, S. (2021). Two Opposing Functions of Angiotensin-Converting Enzyme (ACE) That Links Hypertension, Dementia, and Aging. International Journal of Molecular Sciences, 22(24), 13178. https://doi.org/10.3390/ijms222413178