
Differential Actions of Muscarinic Receptor Subtypes in Gastric, Pancreatic, and Colon Cancer

 , ,
, , 

Abstract
1. Introduction
2. Muscarinic Receptor Ligands
2.1. Overview
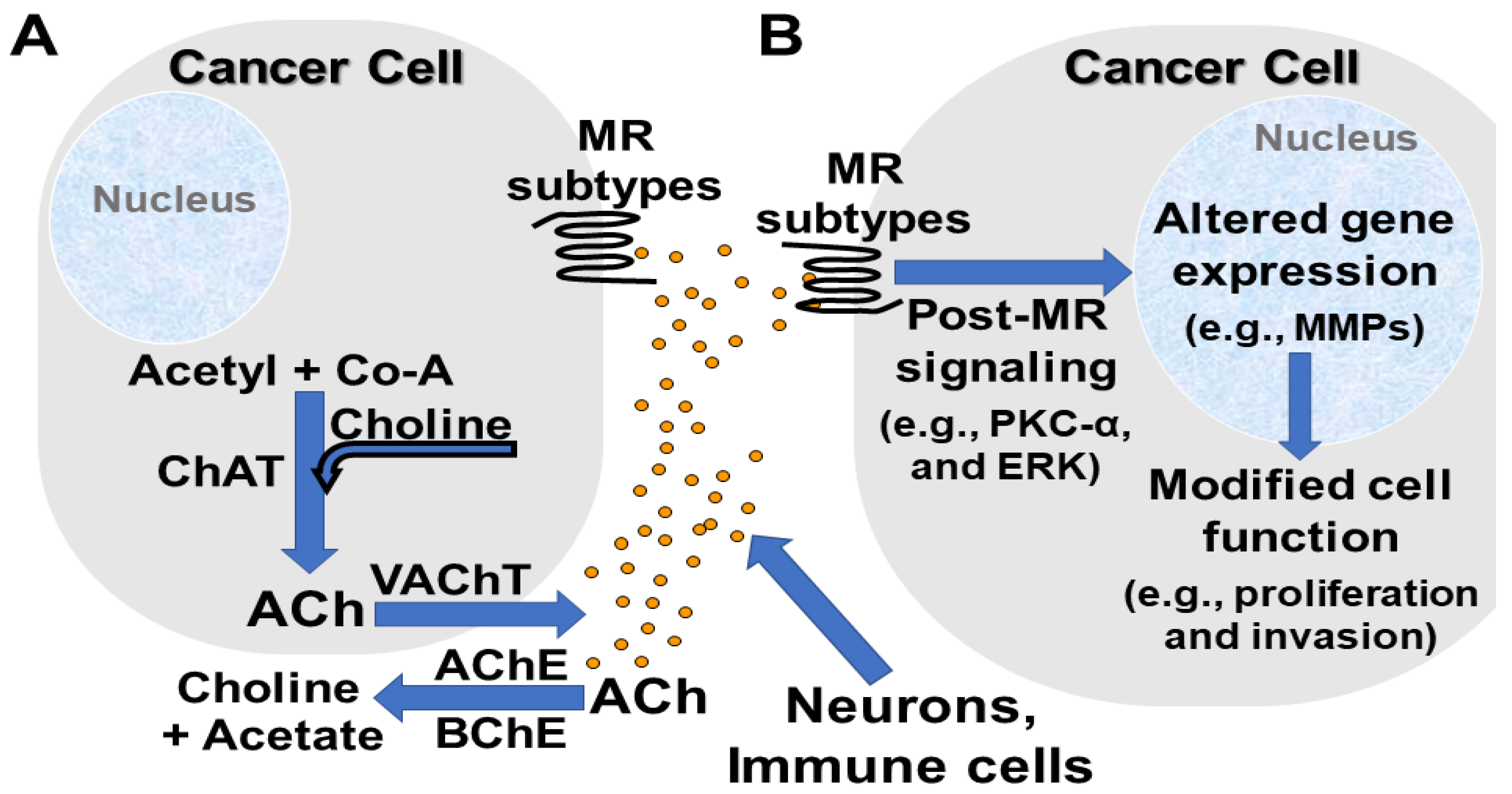
2.2. Neuronal and Non-Neuronal Acetylcholine
2.3. Bile Acids as Physiological Bioactive Muscarinic Receptor Ligands
3. Muscarinic Receptor Subtypes, Signaling, and Anatomic and Cellular Distribution
3.1. Overview
3.2. CHRM1/M1R
3.3. CHRM2/M2R
3.4. CHRM3/M3R
3.5. CHRM4/M4R
3.6. CHRM5/M5R
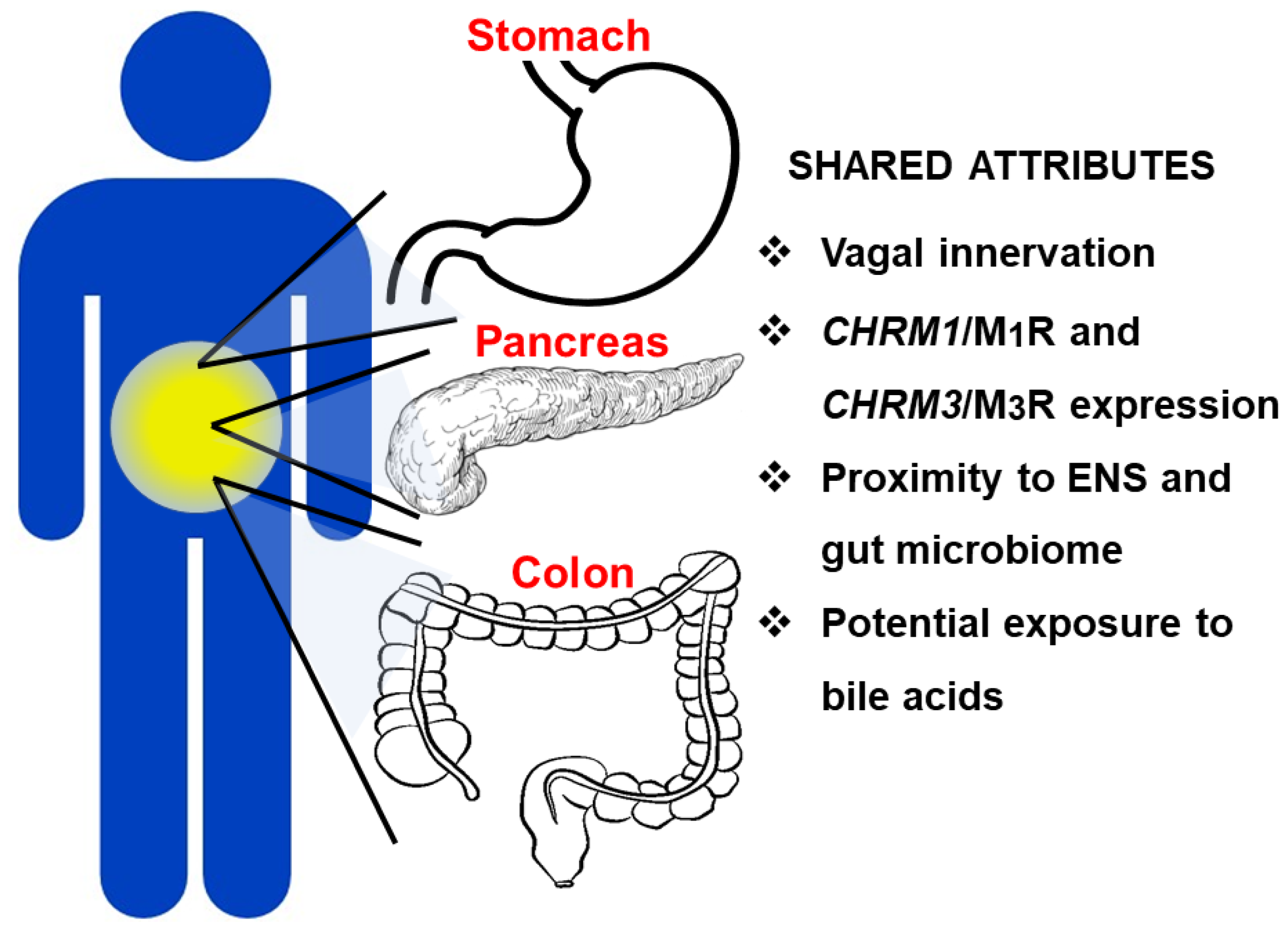
4. Differential Role of Muscarinic Receptor Subtype Activation in GI Cancers
4.1. Overview
4.2. Gastric Adenocarcinoma
4.3. Pancreatic Adenocarcinoma
4.4. Colon Adenocarcinoma
4.5. Role of Muscarinic Receptor Activation within the Enteric Nervous System in GI Neoplasia
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schledwitz, A.; Xie, G.; Raufman, J.P. Exploiting unique features of the gut-brain interface to combat gastrointestinal cancer. J. Clin. Investig. 2021, 131, e143776. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D.; et al. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H. Checks and balances on cholinergic signaling in brain and body function. Trends Neurosci. 2015, 38, 448–458. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.L.; Torre, L.A.; Soerjomataram, I.; Hayes, R.B.; Bray, F.; Weber, T.K.; Jemal, A. Global patterns and trends in colorectal cancer incidence in young adults. Gut 2019, 68, 2179–2185. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; Davis, R.J.; Nomikos, G.; Raufman, J.P. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G591–G597. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Fujii, T. Basic and clinical aspects of non-neuronal acetylcholine: Overview of non-neuronal cholinergic systems and their biological significance. J. Pharmacol. Sci. 2008, 106, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.; Lips, K.S. The non-neuronal cholinergic system in health and disease. Pharmacology 2013, 92, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.P. Muscarinic receptors and ligands in cancer. Am. J. Physiol. Cell Physiol. 2009, 296, C221–C232. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; Yang, L.; Shulkes, A.; Vonlaufen, A.; Poljak, A.; Bustamante, S.; Warren, A.; Xu, Z.; Guilhaus, M.; Pirola, R.; et al. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17397–17402. [Google Scholar] [CrossRef]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Expression and Function of the Cholinergic System in Immune Cells. Front. Immunol. 2017, 8, 1085. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Grafe, F.; Wohlrab, W.; Neubert, R.H.; Brandsch, M. Functional characterization of a high-affinity choline transport system in human keratinocytes. J. Investig. Dermatol. 2002, 119, 118–121. [Google Scholar] [CrossRef]
- Gramatyka, M.; Skorupa, A.; Sokol, M. Nuclear magnetic resonance spectroscopy reveals metabolic changes in living cardiomyocytes after low doses of ionizing radiation. Acta Biochim. Pol. 2018, 65, 309–318. [Google Scholar] [CrossRef]
- Rabkin, S.W.; Cheng, K.M. A genetic abnormality of cardiac myocytes from the blind mutant (RC) chick heart: Abnormalities of cardiac structure and choline transport. Basic Res. Cardiol. 1992, 87, 610–617. [Google Scholar] [CrossRef]
- Sassa, N.; Kato, K.; Abe, S.; Iwano, S.; Ito, S.; Ikeda, M.; Shimamoto, K.; Yamamoto, S.; Yamamoto, T.; Gotoh, M.; et al. Evaluation of 11C-choline PET/CT for primary diagnosis and staging of urothelial carcinoma of the upper urinary tract: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2232–2241. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, F.C. The distribution of acetylcholine in the peripheral and the central nervous system. J. Physiol. 1941, 99, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Woolf, N.J.; Butcher, L.L. Cholinergic systems in the rat brain: III. Projections from the pontomesencephalic tegmentum to the thalamus, tectum, basal ganglia, and basal forebrain. Brain Res. Bull. 1986, 16, 603–637. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdes-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Reardon, C.; Duncan, G.S.; Brustle, A.; Brenner, D.; Tusche, M.W.; Olofsson, P.S.; Rosas-Ballina, M.; Tracey, K.J.; Mak, T.W. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 1410–1415. [Google Scholar] [CrossRef]
- Jiang, W.; Li, D.; Han, R.; Zhang, C.; Jin, W.N.; Wood, K.; Liu, Q.; Shi, F.D.; Hao, J. Acetylcholine-producing NK cells attenuate CNS inflammation via modulation of infiltrating monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E6202–E6211. [Google Scholar] [CrossRef]
- Sastry, B.V. Human placental cholinergic system. Biochem. Pharmacol. 1997, 53, 1577–1586. [Google Scholar] [CrossRef]
- Grando, S.A.; Kist, D.A.; Qi, M.; Dahl, M.V. Human keratinocytes synthesize, secrete, and degrade acetylcholine. J. Investig. Dermatol. 1993, 101, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Fields, W.C.; Rocha-Resende, C.; Resende, R.R.; Guatimosim, S.; Prado, V.F.; Gros, R.; Prado, M.A. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. 2013, 27, 5072–5082. [Google Scholar] [CrossRef]
- Proskocil, B.J.; Sekhon, H.S.; Jia, Y.; Savchenko, V.; Blakely, R.D.; Lindstrom, J.; Spindel, E.R. Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 2004, 145, 2498–2506. [Google Scholar] [CrossRef]
- Kirkpatrick, C.J.; Bittinger, F.; Nozadze, K.; Wessler, I. Expression and function of the non-neuronal cholinergic system in endothelial cells. Life Sci. 2003, 72, 2111–2116. [Google Scholar] [CrossRef]
- Hanna-Mitchell, A.T.; Beckel, J.M.; Barbadora, S.; Kanai, A.J.; de Groat, W.C.; Birder, L.A. Non-neuronal acetylcholine and urinary bladder urothelium. Life Sci. 2007, 80, 2298–2302. [Google Scholar] [CrossRef] [PubMed]
- Ockenga, W.; Kuhne, S.; Bocksberger, S.; Banning, A.; Tikkanen, R. Non-neuronal functions of the m2 muscarinic acetylcholine receptor. Genes 2013, 4, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xia, H.; Tang, Q.; Xu, H.; Wei, G.; Chen, Y.; Dai, X.; Gong, Q.; Bi, F. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci. Rep. 2017, 7, 40802. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Sekhon, H.S.; Jia, Y.; Keller, J.A.; Blusztajn, J.K.; Mark, G.P.; Spindel, E.R. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res. 2003, 63, 214–221. [Google Scholar]
- Cheng, K.; Chen, Y.; Zimniak, P.; Raufman, J.P.; Xiao, Y.; Frucht, H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim. Biophys. Acta 2002, 1588, 48–55. [Google Scholar] [CrossRef]
- Cheng, K.; Raufman, J.P. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem. Pharmacol. 2005, 70, 1035–1047. [Google Scholar] [CrossRef]
- Shant, J.; Cheng, K.; Marasa, B.S.; Wang, J.Y.; Raufman, J.P. Akt-dependent NF-kappaB activation is required for bile acids to rescue colon cancer cells from stress-induced apoptosis. Exp. Cell Res. 2009, 315, 432–450. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Jakubik, J.; El-Fakahany, E.E. Allosteric Modulation of GPCRs of Class A by Cholesterol. Int. J. Mol. Sci. 2021, 22, 1953. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Dawson, P.A.; Rao, A.; Drachenberg, C.B.; Heath, J.; Shang, A.C.; Hu, S.; Zhan, M.; Polli, J.E.; Cheng, K. Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis 2015, 36, 1193–1200. [Google Scholar] [CrossRef]
- Cheng, K.; Metry, M.; Felton, J.; Shang, A.C.; Drachenberg, C.B.; Xu, S.; Zhan, M.; Schumacher, J.; Guo, G.L.; Polli, J.E.; et al. Diminished gallbladder filling, increased fecal bile acids, and promotion of colon epithelial cell proliferation and neoplasia in fibroblast growth factor 15-deficient mice. Oncotarget 2018, 9, 25572–25585. [Google Scholar] [CrossRef]
- Raufman, J.P.; Cheng, K.; Zimniak, P. Activation of muscarinic receptor signaling by bile acids: Physiological and medical implications. Dig. Dis. Sci. 2003, 48, 1431–1444. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Zimniak, P.; Bartoszko-Malik, A. Lithocholyltaurine interacts with cholinergic receptors on dispersed chief cells from guinea pig stomach. Am. J. Physiol. 1998, 274, G997–G1004. [Google Scholar] [CrossRef]
- Sheikh Abdul Kadir, S.H.; Miragoli, M.; Abu-Hayyeh, S.; Moshkov, A.V.; Xie, Q.; Keitel, V.; Nikolaev, V.O.; Williamson, C.; Gorelik, J. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS ONE 2010, 5, e9689. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Xie, G.; Raufman, J.P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human cecal bile acids: Concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef]
- Cheng, K.; Xie, G.; Raufman, J.P. Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem. Pharmacol. 2007, 73, 1001–1012. [Google Scholar] [CrossRef]
- Said, A.H.; Hu, S.; Abutaleb, A.; Watkins, T.; Cheng, K.; Chahdi, A.; Kuppusamy, P.; Saxena, N.; Xie, G.; Raufman, J.P. Interacting post-muscarinic receptor signaling pathways potentiate matrix metalloproteinase-1 expression and invasion of human colon cancer cells. Biochem. J. 2017, 474, 647–665. [Google Scholar] [CrossRef]
- Pedersen, J.E.; Bergqvist, C.A.; Larhammar, D. Evolution of the Muscarinic Acetylcholine Receptors in Vertebrates. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic Signaling via Muscarinic Receptors Directly and Indirectly Suppresses Pancreatic Tumorigenesis and Cancer Stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Drachenberg, C.; Yamada, M.; Wess, J.; Raufman, J.P. Cholinergic agonist-induced pepsinogen secretion from murine gastric chief cells is mediated by M1 and M3 muscarinic receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G521–G529. [Google Scholar] [CrossRef]
- Aihara, T.; Nakamura, Y.; Taketo, M.M.; Matsui, M.; Okabe, S. Cholinergically stimulated gastric acid secretion is mediated by M(3) and M(5) but not M(1) muscarinic acetylcholine receptors in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1199–G1207. [Google Scholar] [CrossRef]
- Iino, S.; Nojyo, Y. Muscarinic M(2) acetylcholine receptor distribution in the guinea-pig gastrointestinal tract. Neuroscience 2006, 138, 549–559. [Google Scholar] [CrossRef]
- Spencer, D.G., Jr.; Horvath, E.; Traber, J. Direct autoradiographic determination of M1 and M2 muscarinic acetylcholine receptor distribution in the rat brain: Relation to cholinergic nuclei and projections. Brain Res. 1986, 380, 59–68. [Google Scholar] [CrossRef]
- Takeuchi, T.; Fujinami, K.; Goto, H.; Fujita, A.; Taketo, M.M.; Manabe, T.; Matsui, M.; Hata, F. Roles of M2 and M4 muscarinic receptors in regulating acetylcholine release from myenteric neurons of mouse ileum. J. Neurophysiol. 2005, 93, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Endoh, T.; Hayashi, S.; Aihara, T. Activation of Muscarinic Acetylcholine Receptor Subtype 4 Is Essential for Cholinergic Stimulation of Gastric Acid Secretion: Relation to D Cell/Somatostatin. Front. Pharmacol. 2016, 7, 278. [Google Scholar] [CrossRef]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef]
- Wang, L.; Xu, J.; Xia, Y.; Yin, K.; Li, Z.; Li, B.; Wang, W.; Xu, H.; Yang, L.; Xu, Z. Muscarinic acetylcholine receptor 3 mediates vagus nerve-induced gastric cancer. Oncogenesis 2018, 7, 88. [Google Scholar] [CrossRef]
- Zhang, L.; Xiu, D.; Zhan, J.; He, X.; Guo, L.; Wang, J.; Tao, M.; Fu, W.; Zhang, H. High expression of muscarinic acetylcholine receptor 3 predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Onco. Targets Ther. 2016, 9, 6719–6726. [Google Scholar] [CrossRef]
- Delvalle, N.M.; Fried, D.E.; Rivera-Lopez, G.; Gaudette, L.; Gulbransen, B.D. Cholinergic activation of enteric glia is a physiological mechanism that contributes to the regulation of gastrointestinal motility. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G473–G483. [Google Scholar] [CrossRef]
- Hering, N.A.; Liu, V.; Kim, R.; Weixler, B.; Droeser, R.A.; Arndt, M.; Pozios, I.; Beyer, K.; Kreis, M.E.; Seeliger, H. Blockage of Cholinergic Signaling via Muscarinic Acetylcholine Receptor 3 Inhibits Tumor Growth in Human Colorectal Adenocarcinoma. Cancers 2021, 13, 3220. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Wilson, J.M.; Remke, D.H.; Mahmood, M.S.; Uddin, M.J.; Wess, J.; Patel, S.; Marnett, L.J.; Niswender, C.M.; Jones, C.K.; et al. Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release. Neuron 2016, 91, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Saternos, H.C.; Almarghalani, D.A.; Gibson, H.M.; Meqdad, M.A.; Antypas, R.B.; Lingireddy, A.; AbouAlaiwi, W.A. Distribution and function of the muscarinic receptor subtypes in the cardiovascular system. Physiol. Genom. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Foster, D.J.; Gentry, P.R.; Lizardi-Ortiz, J.E.; Bridges, T.M.; Wood, M.R.; Niswender, C.M.; Sulzer, D.; Lindsley, C.W.; Xiang, Z.; Conn, P.J. M5 receptor activation produces opposing physiological outcomes in dopamine neurons depending on the receptor’s location. J. Neurosci. 2014, 34, 3253–3262. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Loose, M.D.; Qi, M.; Levey, A.I.; Hille, B.; McKnight, G.S.; Idzerda, R.L.; Nathanson, N.M. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13311–13316. [Google Scholar] [CrossRef] [PubMed]
- Abrams, P.; Andersson, K.E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Anagnostaras, S.G.; Murphy, G.G.; Hamilton, S.E.; Mitchell, S.L.; Rahnama, N.P.; Nathanson, N.M.; Silva, A.J. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat. Neurosci. 2003, 6, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.J.; Sotnikova, T.D.; Gainetdinov, R.R.; Huang, S.Y.; Caron, M.G.; Tonegawa, S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 15312–15317. [Google Scholar] [CrossRef] [PubMed]
- Hardouin, S.N.; Richmond, K.N.; Zimmerman, A.; Hamilton, S.E.; Feigl, E.O.; Nathanson, N.M. Altered cardiovascular responses in mice lacking the M(1) muscarinic acetylcholine receptor. J. Pharmacol. Exp. Ther. 2002, 301, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Kirchner, D.; Kunze, F.; Chretien, E.M.; Michel-Reher, M.B.; Voigt, N.; Knaut, M.; Michel, M.C.; Ravens, U.; Dobrev, D. Muscarinic type-1 receptors contribute to IK,ACh in human atrial cardiomyocytes and are upregulated in patients with chronic atrial fibrillation. Int. J. Cardiol. 2018, 255, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Anisuzzaman, A.S.; Morishima, S.; Suzuki, F.; Tanaka, T.; Yoshiki, H.; Sathi, Z.S.; Akino, H.; Yokoyama, O.; Muramatsu, I. Assessment of muscarinic receptor subtypes in human and rat lower urinary tract by tissue segment binding assay. J. Pharmacol. Sci. 2008, 106, 271–279. [Google Scholar] [CrossRef]
- Witte, L.P.; Chapple, C.R.; de la Rosette, J.J.; Michel, M.C. Cholinergic innervation and muscarinic receptors in the human prostate. Eur. Urol. 2008, 54, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.Q.; Xu, L.H.; Zhang, M.; Xu, C. Muscarinic acetylcholine receptor M1 mediates prostate cancer cell migration and invasion through hedgehog signaling. Asian J. Androl. 2018, 20, 608–614. [Google Scholar] [CrossRef]
- Mannan Baig, A.; Khan, N.A.; Effendi, V.; Rana, Z.; Ahmad, H.R.; Abbas, F. Differential receptor dependencies: Expression and significance of muscarinic M1 receptors in the biology of prostate cancer. Anticancer Drugs 2017, 28, 75–87. [Google Scholar] [CrossRef]
- Gautam, D.; Duttaroy, A.; Cui, Y.; Han, S.J.; Deng, C.; Seeger, T.; Alzheimer, C.; Wess, J. M1-M3 muscarinic acetylcholine receptor-deficient mice: Novel phenotypes. J. Mol. Neurosci. 2006, 30, 157–160. [Google Scholar] [CrossRef]
- Ryberg, A.T.; Warfvinge, G.; Axelsson, L.; Soukup, O.; Gotrick, B.; Tobin, G. Expression of muscarinic receptor subtypes in salivary glands of rats, sheep and man. Arch. Oral Biol. 2008, 53, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Han, S.J.; Heard, T.S.; Cui, Y.; Miller, G.; Bloodworth, L.; Wess, J. Cholinergic stimulation of amylase secretion from pancreatic acinar cells studied with muscarinic acetylcholine receptor mutant mice. J. Pharmacol. Exp. Ther. 2005, 313, 995–1002. [Google Scholar] [CrossRef]
- Cheng, K.; Xie, G.; Khurana, S.; Heath, J.; Drachenberg, C.B.; Timmons, J.; Shah, N.; Raufman, J.P. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol. Cancer 2014, 13, 77. [Google Scholar] [CrossRef]
- Castro, J.M.; Resende, R.R.; Mirotti, L.; Florsheim, E.; Albuquerque, L.L.; Lino-dos-Santos-Franco, A.; Gomes, E.; de Lima, W.T.; de Franco, M.; Ribeiro, O.G.; et al. Role of m2 muscarinic receptor in the airway response to methacholine of mice selected for minimal or maximal acute inflammatory response. Biomed. Res. Int. 2013, 2013, 805627. [Google Scholar] [CrossRef] [PubMed]
- Schlenz, H.; Kummer, W.; Jositsch, G.; Wess, J.; Krasteva, G. Muscarinic receptor-mediated bronchoconstriction is coupled to caveolae in murine airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L626–L636. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Bharucha, A.E.; Thomforde, G.M.; Kost, L.J.; Phillips, S.F. Model of rapid gastrointestinal transit in dogs: Effects of muscarinic antagonists and a nitric oxide synthase inhibitor. Neurogastroenterol. Motil. 2002, 14, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M.; Harris, G.C. Selective inactivation of muscarinic M2 and M3 receptors in guinea-pig ileum and atria in vitro. Br. J. Pharmacol. 1993, 109, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Luthin, G.R.; Ruggieri, M.R. Muscarinic acetylcholine receptor subtypes mediating urinary bladder contractility and coupling to GTP binding proteins. J. Pharmacol. Exp. Ther. 1995, 273, 959–966. [Google Scholar] [PubMed]
- Choppin, A.; Eglen, R.M.; Hegde, S.S. Pharmacological characterization of muscarinic receptors in rabbit isolated iris sphincter muscle and urinary bladder smooth muscle. Br. J. Pharmacol. 1998, 124, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Gil, D.W.; Krauss, H.A.; Bogardus, A.M.; WoldeMussie, E. Muscarinic receptor subtypes in human iris-ciliary body measured by immunoprecipitation. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1434–1442. [Google Scholar]
- Roffel, A.F.; Elzinga, C.R.; Zaagsma, J. Muscarinic M3 receptors mediate contraction of human central and peripheral airway smooth muscle. Pulm. Pharmacol. 1990, 3, 47–51. [Google Scholar] [CrossRef]
- Beny, J.L.; Nguyen, M.N.; Marino, M.; Matsui, M. Muscarinic receptor knockout mice confirm involvement of M3 receptor in endothelium-dependent vasodilatation in mouse arteries. J. Cardiovasc. Pharmacol. 2008, 51, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Chacon, I.; Xie, G.; Yamada, M.; Wess, J.; Raufman, J.P.; Kennedy, R.H. Vasodilatory effects of cholinergic agonists are greatly diminished in aorta from M3R-/- mice. Eur. J. Pharmacol. 2004, 493, 127–132. [Google Scholar] [CrossRef]
- Ehrenkaufer, R.L.; Klam, S.; Makoroff, K.; Giandinoto, S.; Morton, T.; Moroney, D.; Nowak, P. Internal-surface reversed-phase chromatography for plasma metabolite analysis of radiopharmaceuticals. Int. J. Rad. Appl. Instrum. B 1992, 19, 651–657. [Google Scholar] [CrossRef]
- Wan, J.; Wang, J.; Wagner Ii, L.E.; Wang, O.H.; Gui, F.; Chen, J.; Zhu, X.; Haddock, A.N.; Edenfield, B.H.; Haight, B.; et al. Pancreatic specific CHRM3 activation causes pancreatitis in mice. JCI Insight 2021, 6, e132585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Rossi, M.; Cohen, A.; Pham, J.; Zheng, H.; Dattaroy, D.; Mukaibo, T.; Melvin, J.E.; Langel, J.L.; Hattar, S.; et al. Allosteric modulation of beta-cell M3 muscarinic acetylcholine receptors greatly improves glucose homeostasis in lean and obese mice. Proc. Natl. Acad. Sci. USA 2019, 116, 18684–18690. [Google Scholar] [CrossRef] [PubMed]
- Scully, C. Drug effects on salivary glands: Dry mouth. Oral Dis. 2003, 9, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Gauna, A.E.; Perez, G.; Park, Y.J.; Pauley, K.M.; Kawai, T.; Cha, S. Autoantibodies against muscarinic type 3 receptor in Sjogren’s syndrome inhibit aquaporin 5 trafficking. PLoS ONE 2013, 8, e53113. [Google Scholar] [CrossRef] [PubMed]
- Koo, N.Y.; Li, J.; Hwang, S.M.; Choi, S.Y.; Lee, S.J.; Oh, S.B.; Kim, J.S.; Lee, E.B.; Song, Y.W.; Park, K. Functional epitope of muscarinic type 3 receptor which interacts with autoantibodies from Sjogren’s syndrome patients. Rheumatology 2008, 47, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Frucht, H.; Jensen, R.T.; Dexter, D.; Yang, W.L.; Xiao, Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin. Cancer Res. 1999, 5, 2532–2539. [Google Scholar]
- Van der Westhuizen, E.T.; Choy, K.H.C.; Valant, C.; McKenzie-Nickson, S.; Bradley, S.J.; Tobin, A.B.; Sexton, P.M.; Christopoulos, A. Fine Tuning Muscarinic Acetylcholine Receptor Signaling Through Allostery and Bias. Front. Pharmacol. 2021, 11, 2217. [Google Scholar] [CrossRef] [PubMed]
- Brannan, S.K.; Sawchak, S.; Miller, A.C.; Lieberman, J.A.; Paul, S.M.; Breier, A. Muscarinic Cholinergic Receptor Agonist and Peripheral Antagonist for Schizophrenia. N. Engl. J. Med. 2021, 384, 717–726. [Google Scholar] [CrossRef]
- Wang, L.; Zhi, X.; Zhang, Q.; Wei, S.; Li, Z.; Zhou, J.; Jiang, J.; Zhu, Y.; Yang, L.; Xu, H.; et al. Muscarinic receptor M3 mediates cell proliferation induced by acetylcholine and contributes to apoptosis in gastric cancer. Tumour Biol. 2016, 37, 2105–2117. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Heath, J.; Drachenberg, C.; Raufman, J.P.; Xie, G. Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer 2013, 13, 204. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Shang, A.C.; Drachenberg, C.B.; Zhan, M.; Raufman, J.P. Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget 2017, 8, 21106–21114. [Google Scholar] [CrossRef]
- Felton, J.; Cheng, K.; Shang, A.C.; Hu, S.; Larabee, S.M.; Drachenberg, C.B.; Raufman, J.P. Two sides to colon cancer: Mice mimic human anatomical region disparity in colon cancer development and progression. J. Cancer Metastasis Treat. 2018, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, K.; Shant, J.; Raufman, J.P. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G755–G763. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Shant, J.; Xie, G.; Cheng, K.; Gao, X.M.; Shiu, B.; Shah, N.; Drachenberg, C.B.; Heath, J.; Wess, J.; et al. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis 2011, 32, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Ushiyama, N.; Hosonuma, K.; Suenaga, A.; Kawashima, K. Effects of human antithymocyte globulin on acetylcholine synthesis, its release and choline acetyltransferase transcription in a human leukemic T-cell line. J. Neuroimmunol. 2002, 128, 1–8. [Google Scholar] [CrossRef]
- Amit, M.; Na’ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.M.; Williamson, E.J.; Bassett, J.K.; MacInnis, R.J.; Giles, G.G.; English, D.R. Dietary and biomarker estimates of fatty acids and risk of colorectal cancer. Int. J. Cancer 2015, 137, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef]
- Kruse, A.C.; Li, J.; Hu, J.; Kobilka, B.K.; Wess, J. Novel insights into M3 muscarinic acetylcholine receptor physiology and structure. J. Mol. Neurosci. 2014, 53, 316–323. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef]
- Tan, X.; Sivakumar, S.; Bednarsch, J.; Wiltberger, G.; Kather, J.N.; Niehues, J.; de Vos-Geelen, J.; Valkenburg-van Iersel, L.; Kintsler, S.; Roeth, A.; et al. Nerve fibers in the tumor microenvironment in neurotropic cancer-pancreatic cancer and cholangiocarcinoma. Oncogene 2021, 40, 899–908. [Google Scholar] [CrossRef]
- Duchalais, E.; Guilluy, C.; Nedellec, S.; Touvron, M.; Bessard, A.; Touchefeu, Y.; Bossard, C.; Boudin, H.; Louarn, G.; Neunlist, M.; et al. Colorectal Cancer Cells Adhere to and Migrate Along the Neurons of the Enteric Nervous System. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 31–49. [Google Scholar] [CrossRef]
- Vales, S.; Bacola, G.; Biraud, M.; Touvron, M.; Bessard, A.; Geraldo, F.; Dougherty, K.A.; Lashani, S.; Bossard, C.; Flamant, M.; et al. Tumor cells hijack enteric glia to activate colon cancer stem cells and stimulate tumorigenesis. EBioMedicine 2019, 49, 172–188. [Google Scholar] [CrossRef]
- Sriram, K.; Wiley, S.Z.; Moyung, K.; Gorr, M.W.; Salmeron, C.; Marucut, J.; French, R.P.; Lowy, A.M.; Insel, P.A. Detection and Quantification of GPCR mRNA: An Assessment and Implications of Data from High-Content Methods. ACS Omega 2019, 4, 17048–17059. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Wieland, T.; Tsujimoto, G. How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch. Pharm. 2009, 379, 385–388. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Sources of Acetylcholine | Refs. | |
|---|---|---|
| Neurons | ||
| Autonomic nervous system | Preganglionic sympathetic/parasympathetic neurons | [23] |
| Peripheral nervous system | Terminal ends of axons at neuromuscular junctions | [23] |
| Central nervous system | Primarily interneurons | [23,24] |
| Non-Neuronal Cells | ||
| Immunocytes | CD4 + T cells; B cells; NK cells | [25,26,27] |
| Placental trophoblast | [28] | |
| Keratinocytes | [29] | |
| Cardiomyocytes | [30] | |
| Airway epithelial cells | [31] | |
| Vascular endothelial cells | [32] | |
| Urothelial cells | [33,34] | |
| Cancer cells | Colon, stomach, lung, and others | [12,35,36] |
| MR Subtype (Gene) | G-Protein | Ligands | Tissue Distribution | Physiological Actions | Actions in GI Cancers | Refs. |
|---|---|---|---|---|---|---|
| M1R (CHRM1) | Gq/11 | ACh, BA, cholesterol (allosteric) | Brain, gastric mucosa, respiratory epithelium, skin, melanocytes, immunocytes | Mediates gastric pepsinogen secretion | Protects against PDAC and colon neoplasia | [34,40,41,50,51,52,53] |
| M2R (CHRM2) | Gi/o | ACh | Brain, heart, ENS, gastric mucosa, skin, bladder, melanocytes, smooth muscle, immunocytes | Modulates cardiac rhythm, GI motility | None reported | [34,50,53,54,55,56] |
| M3R (CHRM3) | Gq/11 | ACh, BA | Gastric chief and parietal cells, colon epithelial cells, smooth muscle, ENS, brain, skin, melanocytes, immunocytes | Mediates gastric acid and pepsinogen secretion; GI motility | Promotes gastric and colon cancer cell proliferation and PDAC severity | [37,38,50,52,57,58,59,60,61,62] |
| M4R (CHRM4) | Gi/o | ACh | Brain, gastric mucosa, small intestine, skin, melanocytes, immunocytes | Enhances gastric acid secretion; regulates striatal dopamine release | None reported | [34,50,57,63,64] |
| M5R (CHRM5) | Gq/11 | ACh | Brain, cerebral vasculature, ENS; mRNA expressed in testes, placenta, thyroid, small intestine, immunocytes | Enhances gastric acid secretion; regulates striatal dopamine release; mediates SNc excitability | None reported | [34,50,53,61,63,65,66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schledwitz, A.; Sundel, M.H.; Alizadeh, M.; Hu, S.; Xie, G.; Raufman, J.-P. Differential Actions of Muscarinic Receptor Subtypes in Gastric, Pancreatic, and Colon Cancer. Int. J. Mol. Sci. 2021, 22, 13153. https://doi.org/10.3390/ijms222313153
Schledwitz A, Sundel MH, Alizadeh M, Hu S, Xie G, Raufman J-P. Differential Actions of Muscarinic Receptor Subtypes in Gastric, Pancreatic, and Colon Cancer. International Journal of Molecular Sciences. 2021; 22(23):13153. https://doi.org/10.3390/ijms222313153
Chicago/Turabian StyleSchledwitz, Alyssa, Margaret H. Sundel, Madeline Alizadeh, Shien Hu, Guofeng Xie, and Jean-Pierre Raufman. 2021. "Differential Actions of Muscarinic Receptor Subtypes in Gastric, Pancreatic, and Colon Cancer" International Journal of Molecular Sciences 22, no. 23: 13153. https://doi.org/10.3390/ijms222313153
APA StyleSchledwitz, A., Sundel, M. H., Alizadeh, M., Hu, S., Xie, G., & Raufman, J.-P. (2021). Differential Actions of Muscarinic Receptor Subtypes in Gastric, Pancreatic, and Colon Cancer. International Journal of Molecular Sciences, 22(23), 13153. https://doi.org/10.3390/ijms222313153