
Structural Modelling of KCNQ1 and KCNH2 Double Mutant Proteins, Identified in Two Severe Long QT Syndrome Cases, Reveals New Insights into Cardiac Channelopathies

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
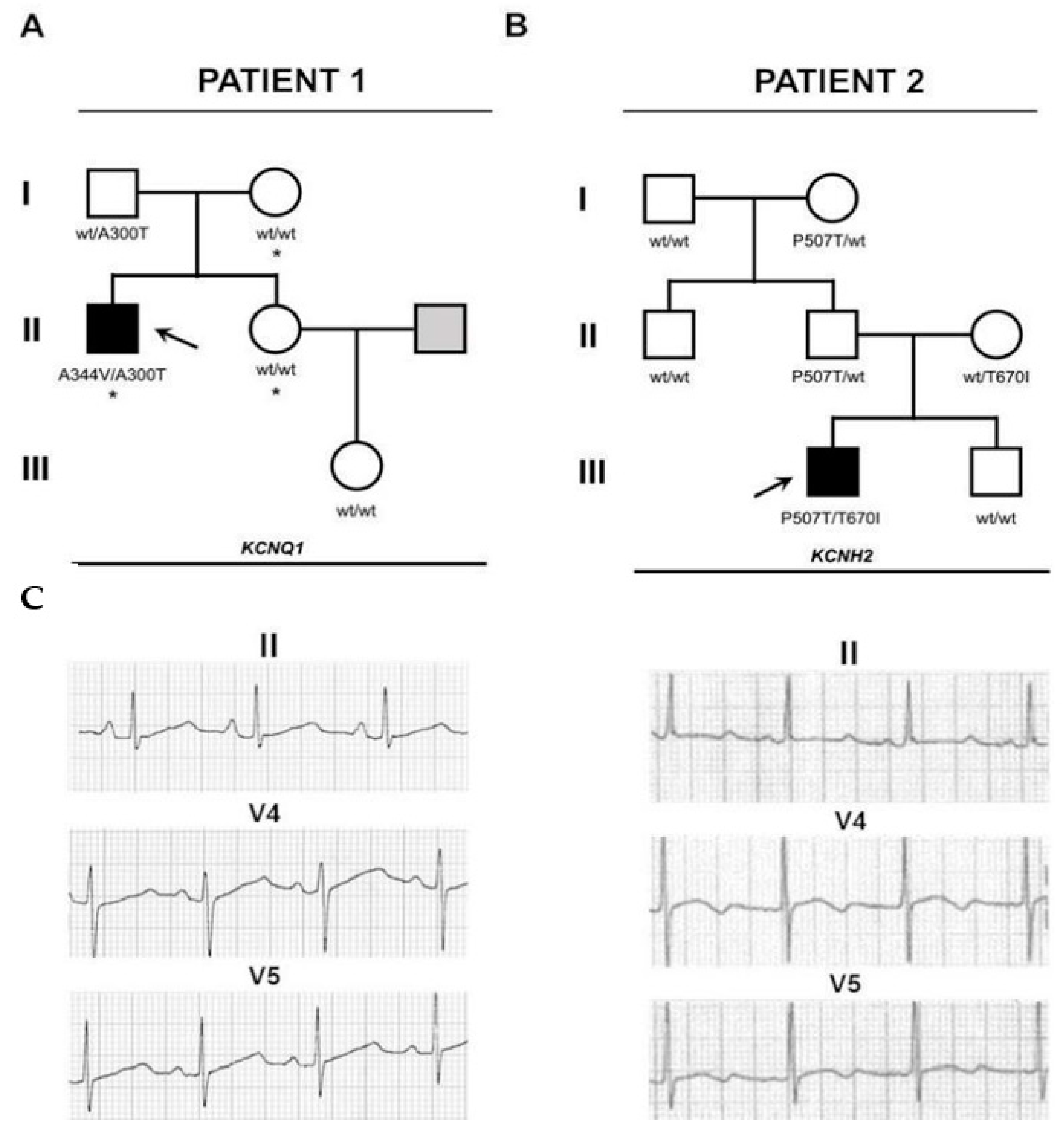
2.1. Sequencing and Variant Analysis
2.1.1. Patient 1
2.1.2. Patient 2
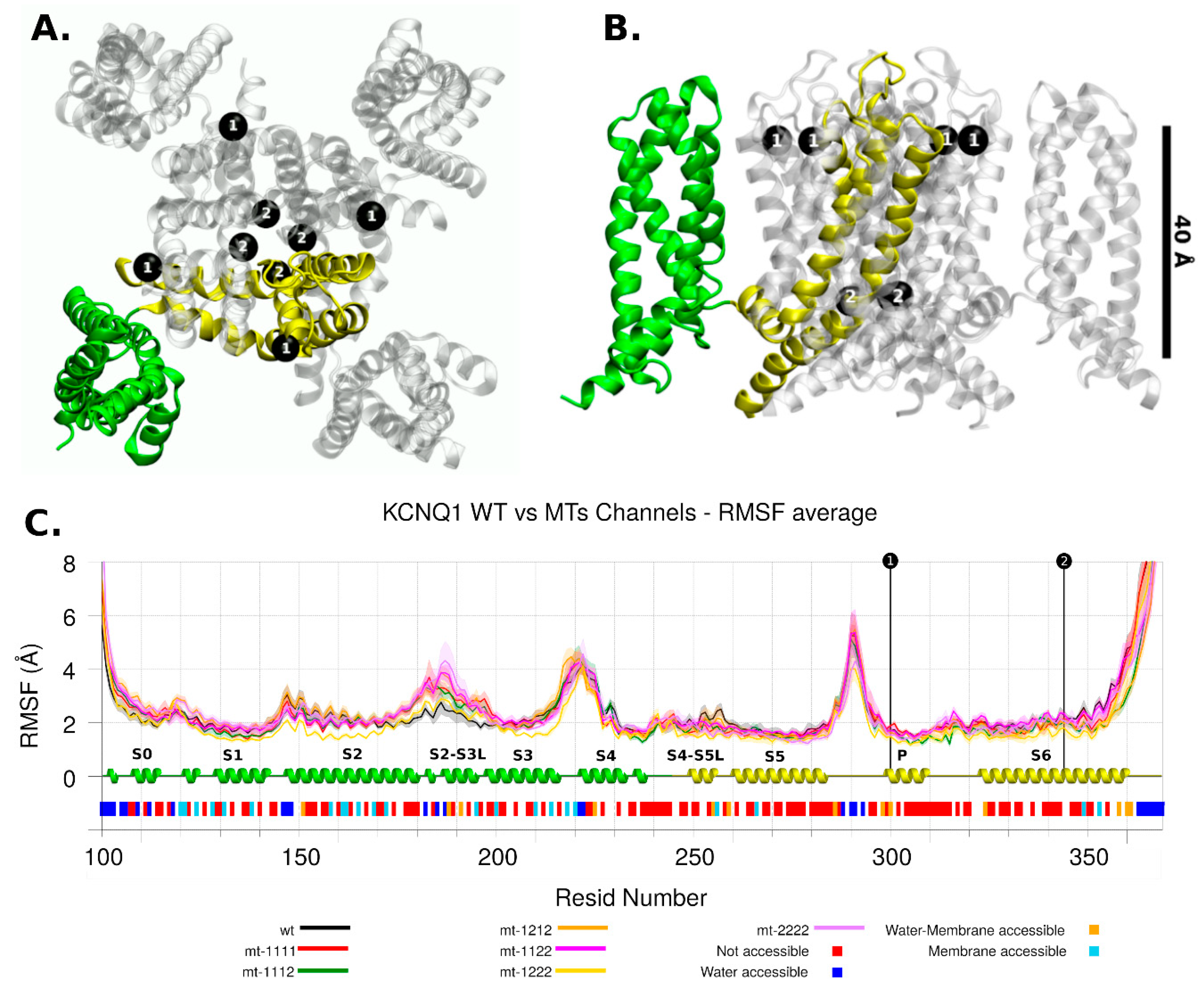
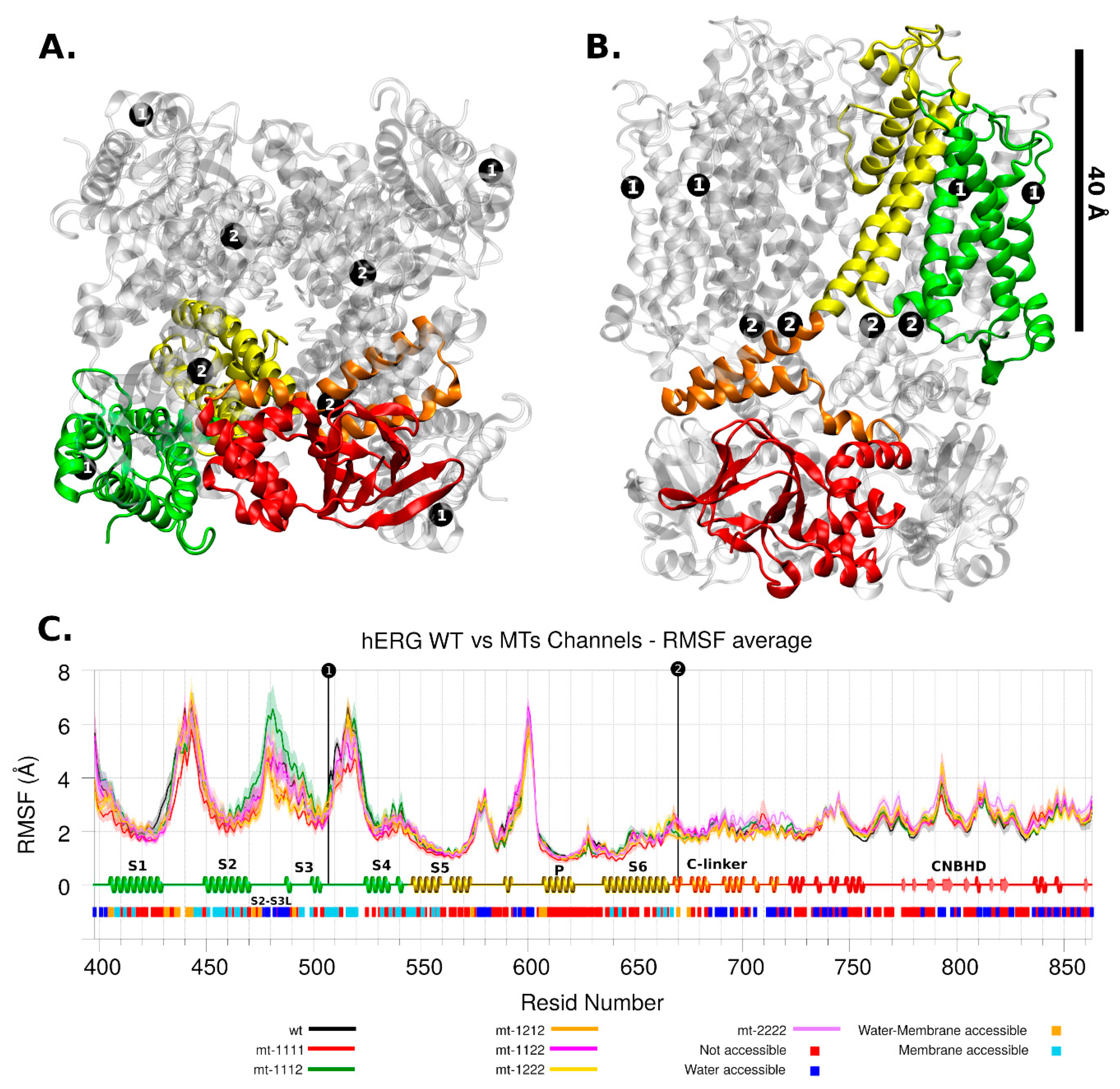
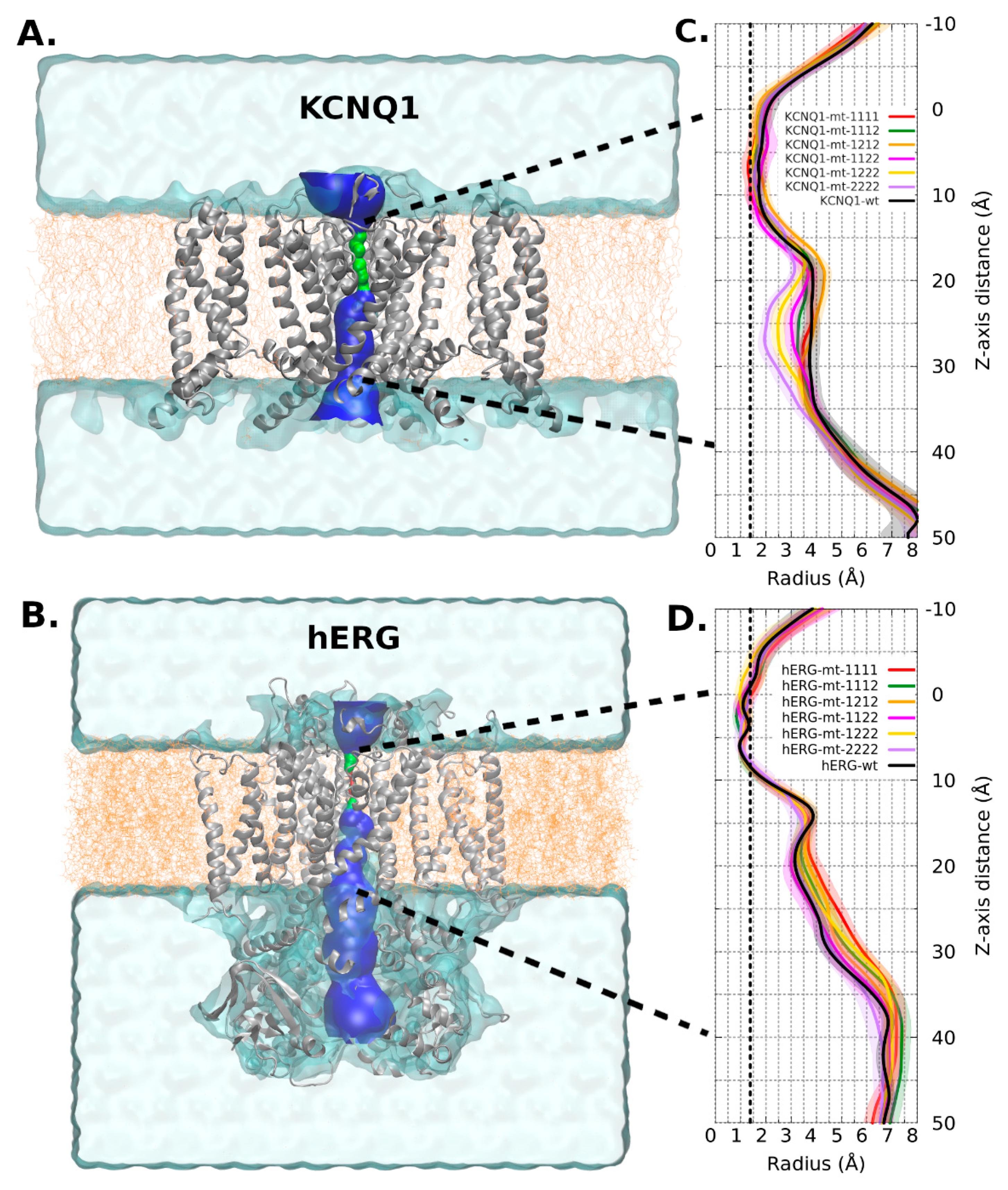
2.2. Molecular Modelling
2.2.1. KCNQ1 Channel
2.2.2. hERG Channel
3. Discussion
4. Materials and Methods
4.1. Subjects
4.1.1. Patient 1
4.1.2. Patient 2
4.2. Molecular Diagnosis
4.2.1. Sequencing and Bioinformatics Analysis of Variants
4.2.2. Homology Models
4.2.3. Secondary Structure Motifs and SASA Analysis
4.3. Molecular Dynamics
4.4. Channel Pore Radius and Volume
4.5. Mutation Free Energy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwartz, P.J.; Ackerman, M.J. The long QT syndrome: A transatlantic clinical approach to diagnosis and therapy. Eur. Heart J. 2013, 34, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, P.J.; Priori, S.G.; Napolitano, C. How Really Rare Are Rare Diseases?: The Intriguing Case of Independent Compound Mutations in the Long QT Syndrome. J. Cardiovasc. Electrophysiol. 2013, 14, 1120–1121. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Wilde, A.A.; Ackerman, M.J. The genetic architecture of long QT syndrome: A critical reappraisal. Trends Cardiovasc. Med. 2018, 28, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, W. Clinical Impact of Genetic Studies in Lethal Inherited Cardiac Arrhythmias. Circ. J. 2008, 72, 1926–1936. [Google Scholar] [CrossRef] [Green Version]
- Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 507–517. [Google Scholar] [CrossRef]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwarz, J.; Towbin, J.; Vincent, M.; et al. Spectrum of mutations in Long-QT Syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, C. Rare cardiac arrhythmias of the pediatric age. II: Syncopal attacks due to paroxysmal ventricular fibrillation (Presentation of 1st case in Italian pediatric literature). Clin. Pediatr. 1963, 45, 656–683. [Google Scholar]
- Itoh, H.; Shimizu, W.; Hayashi, K.; Yamagata, K.; Sakaguchi, T.; Ohno, S.; Makiyama, T.; Akao, M.; Ai, T.; Noda, T.; et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: A Japanese multicenter study. Heart Rhythm 2010, 7, 1411–1418. [Google Scholar] [CrossRef]
- Westenskow, P.; Splawski, I.; Timothy, K.W.; Keating, M.T.; Sanguinetti, M.C. Compound Mutations: A Common Cause of Severe Long-QT Syndrome. Circulation 2004, 109, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Bohnen, M.S.; Peng, G.; Robey, S.H.; Terrenoire, C.; Iyer, V.; Sampson, K.J.; Kass, R.S. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol. Rev. 2017, 97, 89–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Kuenze, G.; Duran, A.M.; Woods, H.; Brewer, K.R.; McDonald, E.F.; Vanoye, C.G.; Alfred, L.G., Jr.; Sanders, C.R.; Meiler, J. Upgraded molecular models of the human KCNQ1 potassium channel. PLoS ONE 2019, 14, e0220415. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-à-go-go -Related K+ Channel hERG. Cell 2017, 169, 422–430.e10. [Google Scholar] [CrossRef] [Green Version]
- Siebrands, C.C.; Binder, S.; Eckhoff, U.; Schmitt, N.; Friederich, P. Long QT 1 mutation KCNQ1 A344V increases local anesthetic sensitivity of the slowly activating delayed rectifier potassium current. Anesthesiology 2006, 105, 511–520. [Google Scholar] [CrossRef]
- Butler, A.S.; Helliwell, M.V.; Zhang, Y.; Hancox, J.C.; Dempsey, C.E. An Update on the Structure of hERG. Front. Pharmacol. 2020, 10, 1572. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 2016, 32, 2936–2946. [Google Scholar] [CrossRef] [Green Version]
- Seebohm, G.; Westenskow, P.; Lang, F.; Sanguinetti, M.C. Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J. Physiol. 2005, 563, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bianchi, L.; Dennis, A.; De Fusco, M.; Brown, A.M.; Casari, G. A Recessive Variant of the Romano-Ward Long-QT Syndrome? Circulation 1998, 97, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Antúnez-Argüelles, E.; Rojo-Domínguez, A.; Arregui-Mena, A.L.; Jacobo-Albavera, L.; Márquez, M.; Iturralde-Torres, P.; Villarreal-Molina, M.T. Compound heterozygous KCNQ1 mutations (A300T/P535T) in a child with sudden unexplained death: Insights into possible molecular mechanisms based on protein modeling. Gene 2017, 627, 40–48. [Google Scholar] [CrossRef]
- Pérez-Pérez, J.M.; Candela, H.; Micol, J.L. Understanding synergy in genetic interactions. Trends Genet. 2009, 25, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.L.; Rodrigues, J.P.; Folkers, G.E.; Boelens, R.; Bonvin, A.M. Proteins feel more than they see: Fine-tuning of binding affinity by properties of the non-interacting surface. J. Mol. Biol. 2014, 426, 2632–2652. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Marcondes, L.; Crawford, J.; Earle, N.; Smith, W.; Hayes, I.; Morrow, P.; Donoghue, T.; Graham, A.; Love, D.; Skinner, J.R.; et al. Long QT molecular autopsy in sudden unexplained death in the young (1–40 years old): Lessons learnt from an eight year experience in New Zealand. PLoS ONE 2018, 13, e0196078. [Google Scholar] [CrossRef] [Green Version]
- Burgos, M.; Arenas, A.; Cabrera, R. Semiconductor Whole Exome Sequencing for the Identification of Genetic Variants in Colombian Patients Clinically Diagnosed with Long QT Syndrome. Mol. Diagn. Ther. 2016, 20, 353–362. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e9. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Smart, O.S.; Neduvelil, J.G.; Wang, X.; Wallace, B.; Sansom, M. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. [Google Scholar] [CrossRef]
- Hutchinson, E.G.; Thornton, J.M. PROMOTIF—A program to identify and analyze structural motifs in proteins. Protein Sci. 1996, 5, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2011, 40, D370–D376. [Google Scholar] [CrossRef]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.J.; Kern, N.R.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.; Holst, O.; et al. CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Larsson, H.P. Insights into Cardiac IKs (KCNQ1/KCNE1) Channels Regulation. Int. J. Mol. Sci. 2020, 21, 9440. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Skjevik, Å.A.; Madej, B.D.; Walker, R.C.; Teigen, K. LIPID11: A Modular Framework for Lipid Simulations Using Amber. J. Phys. Chem. B 2012, 116, 11124–11136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, C.; Xue, L.C.; Roel-Touris, J.; Bonvin, A.M.J.J. Finding the ΔΔ G spot: Are predictors of binding affinity changes upon mutations in protein–protein interactions ready for it? WIREs Comput. Mol. Sci. 2019, 9, e1410. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Kaus, J.W.; Pierce, L.T.; Walker, R.C.; McCammont, J.A. Improving the Efficiency of Free Energy Calculations in the Amber Molecular Dynamics Package. J. Chem. Theory Comput. 2013, 9, 4131–4139. [Google Scholar] [CrossRef] [PubMed]

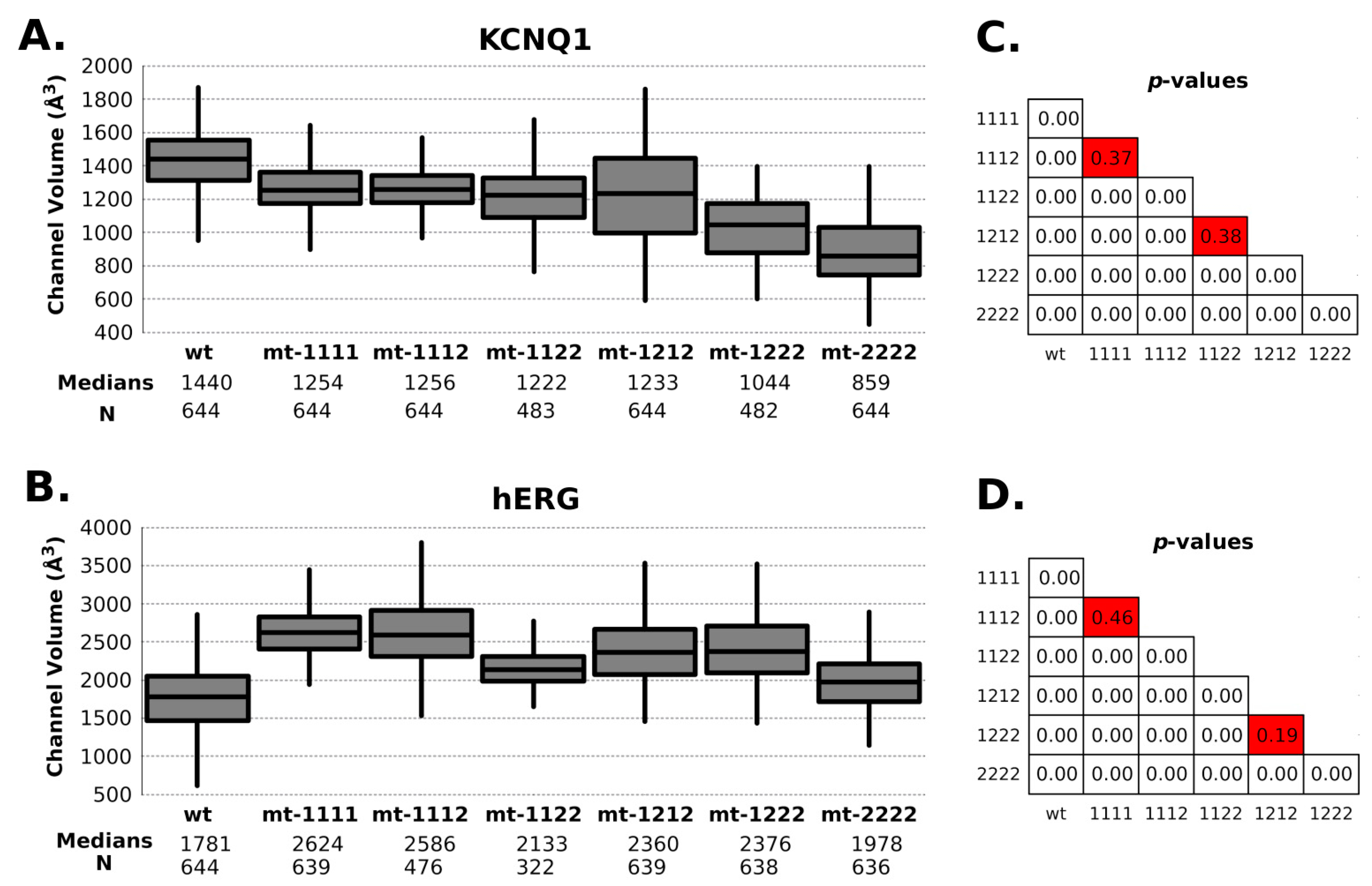

{kind=link}
{kind=link}
{kind=link}
{kind=link}
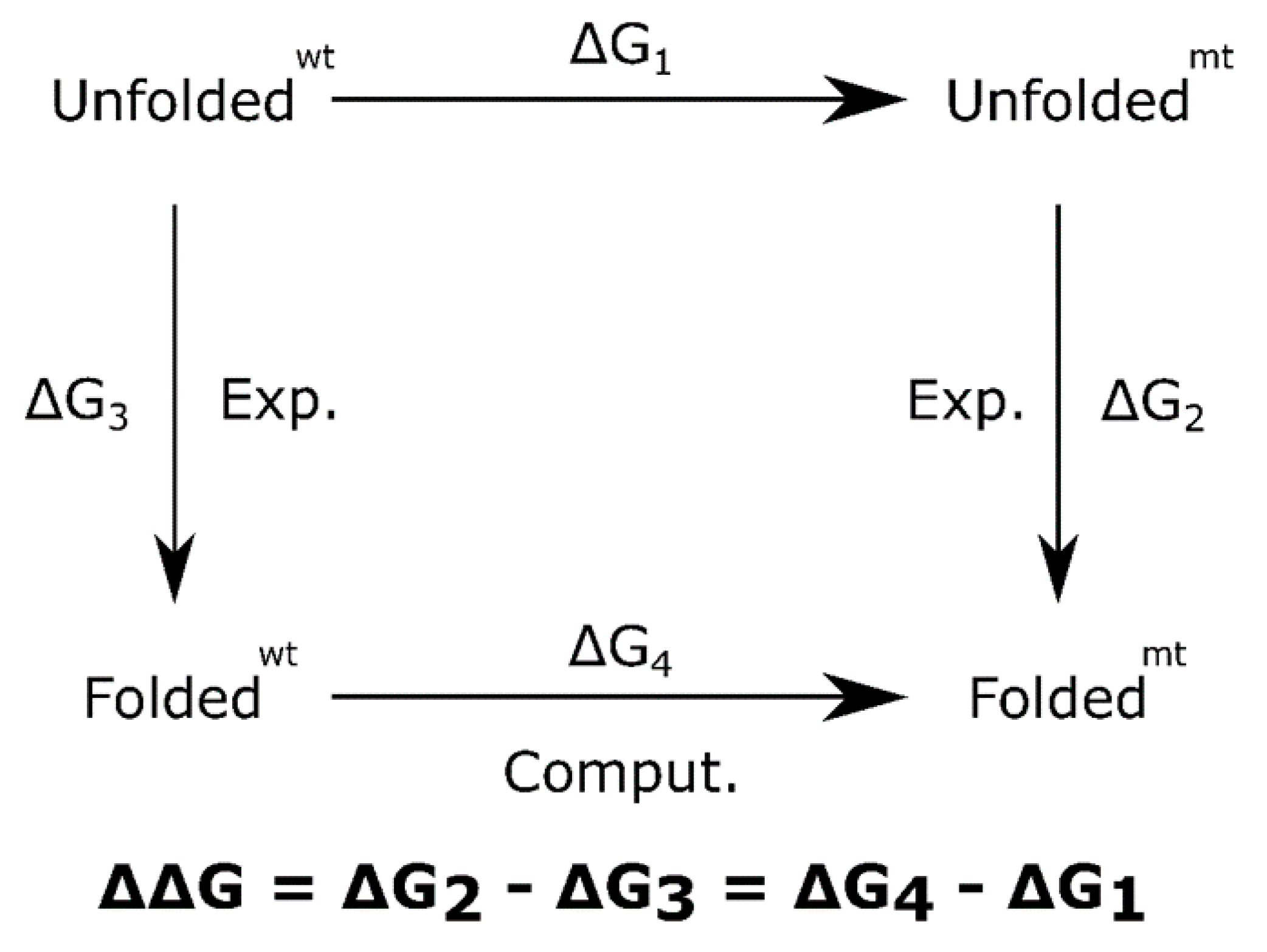
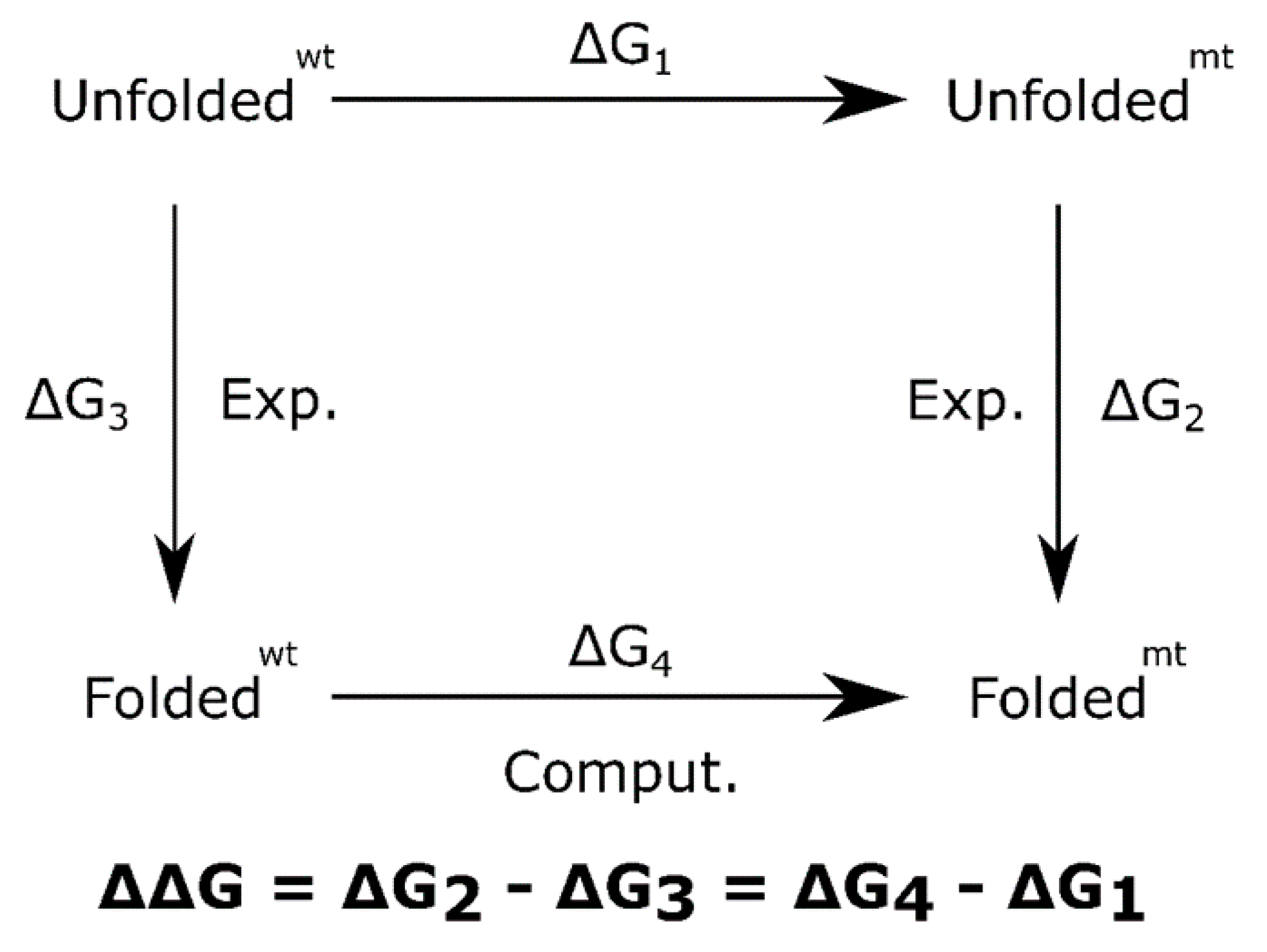
{kind=link}
{kind=link}
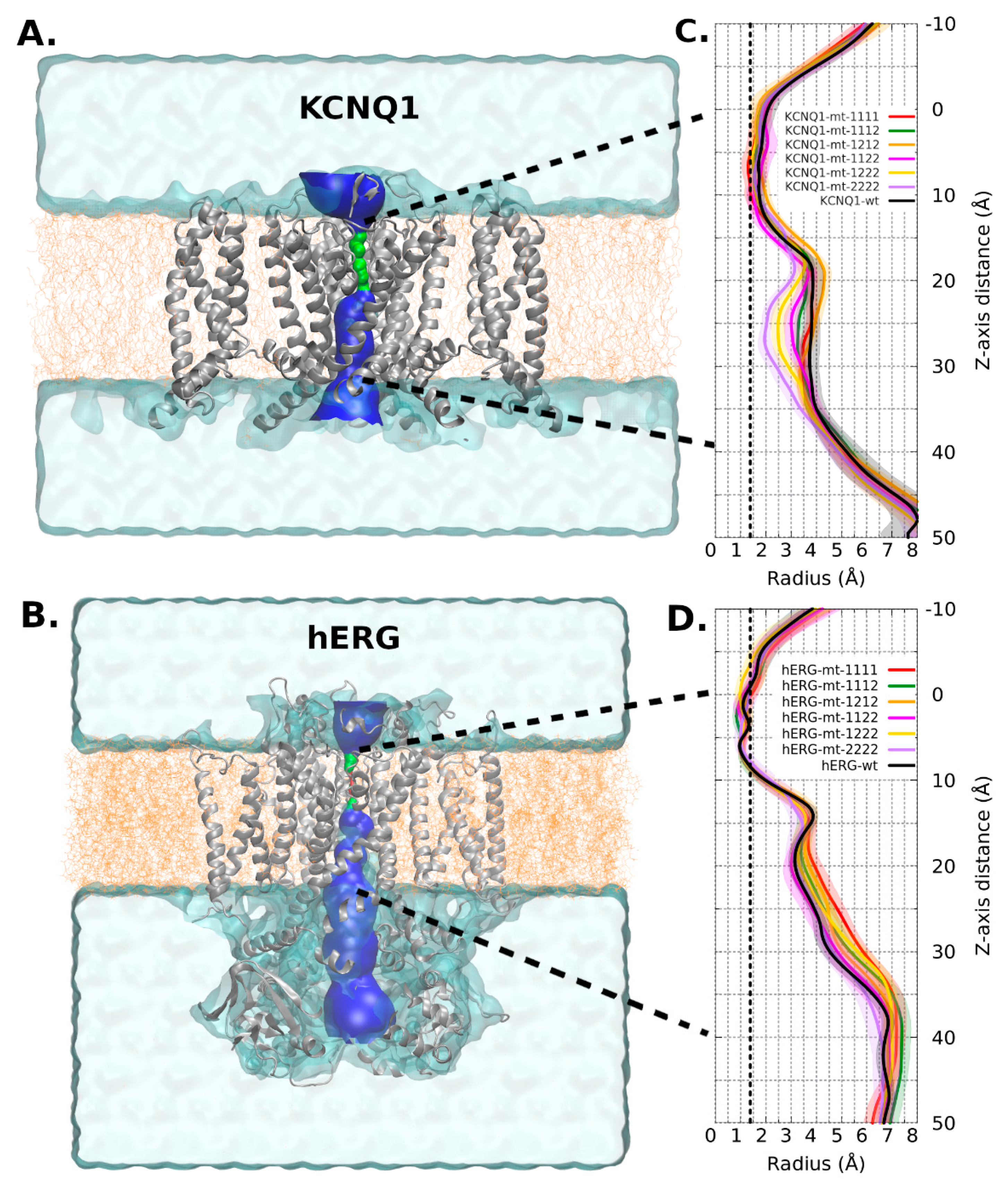
{kind=link}
| ∆∆G (kcal/mol) | |||
|---|---|---|---|
| Channel | Mutant | Monomer | Tetramer |
| KCNQ1 | mt-1111 | 2.56 (0.04) | 10.24 (0.17) |
| mt-2222 | 1.20 (0.04) | 4.81 (0.17) | |
| hERG | mt-1111 | −2.71 (0.05) | −10.83 (0.21) |
| mt-2222 | 1.14 (0.05) | 4.58 (0.19) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agudelo, W.A.; Gil-Quiñones, S.R.; Fonseca, A.; Arenas, A.; Castro, L.; Sierra-Díaz, D.C.; Patarroyo, M.A.; Laissue, P.; Suárez, C.F.; Cabrera, R. Structural Modelling of KCNQ1 and KCNH2 Double Mutant Proteins, Identified in Two Severe Long QT Syndrome Cases, Reveals New Insights into Cardiac Channelopathies. Int. J. Mol. Sci. 2021, 22, 12861. https://doi.org/10.3390/ijms222312861
Agudelo WA, Gil-Quiñones SR, Fonseca A, Arenas A, Castro L, Sierra-Díaz DC, Patarroyo MA, Laissue P, Suárez CF, Cabrera R. Structural Modelling of KCNQ1 and KCNH2 Double Mutant Proteins, Identified in Two Severe Long QT Syndrome Cases, Reveals New Insights into Cardiac Channelopathies. International Journal of Molecular Sciences. 2021; 22(23):12861. https://doi.org/10.3390/ijms222312861
Chicago/Turabian StyleAgudelo, William A., Sebastian Ramiro Gil-Quiñones, Alejandra Fonseca, Alvaro Arenas, Laura Castro, Diana Carolina Sierra-Díaz, Manuel A. Patarroyo, Paul Laissue, Carlos F. Suárez, and Rodrigo Cabrera. 2021. "Structural Modelling of KCNQ1 and KCNH2 Double Mutant Proteins, Identified in Two Severe Long QT Syndrome Cases, Reveals New Insights into Cardiac Channelopathies" International Journal of Molecular Sciences 22, no. 23: 12861. https://doi.org/10.3390/ijms222312861
APA StyleAgudelo, W. A., Gil-Quiñones, S. R., Fonseca, A., Arenas, A., Castro, L., Sierra-Díaz, D. C., Patarroyo, M. A., Laissue, P., Suárez, C. F., & Cabrera, R. (2021). Structural Modelling of KCNQ1 and KCNH2 Double Mutant Proteins, Identified in Two Severe Long QT Syndrome Cases, Reveals New Insights into Cardiac Channelopathies. International Journal of Molecular Sciences, 22(23), 12861. https://doi.org/10.3390/ijms222312861