
Controlled Delivery of Pan-PAD-Inhibitor Cl-Amidine Using Poly(3-Hydroxybutyrate) Microspheres

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. P(3HB) Microsphere Optimization and Characterization
2.2. Surface Morphology of the Cl-Amidine Encapsulated P(3HB) Microspheres Using SEM
2.3. Fourier Transform Infrared Spectroscopy (FTIR) of Cl-Amidine Encapsulated P(3HB) Microspheres
2.4. Porosity of Microspheres
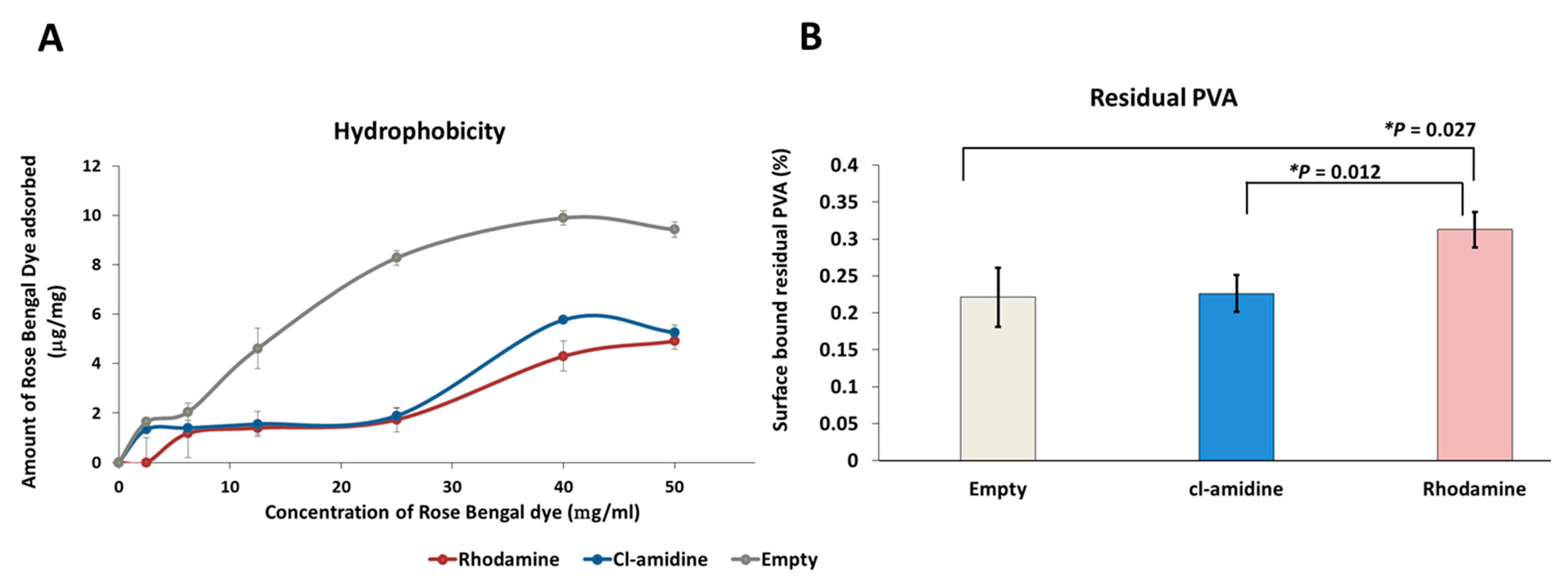
2.5. Surface Hydrophobicity and Residual Percentage PVA
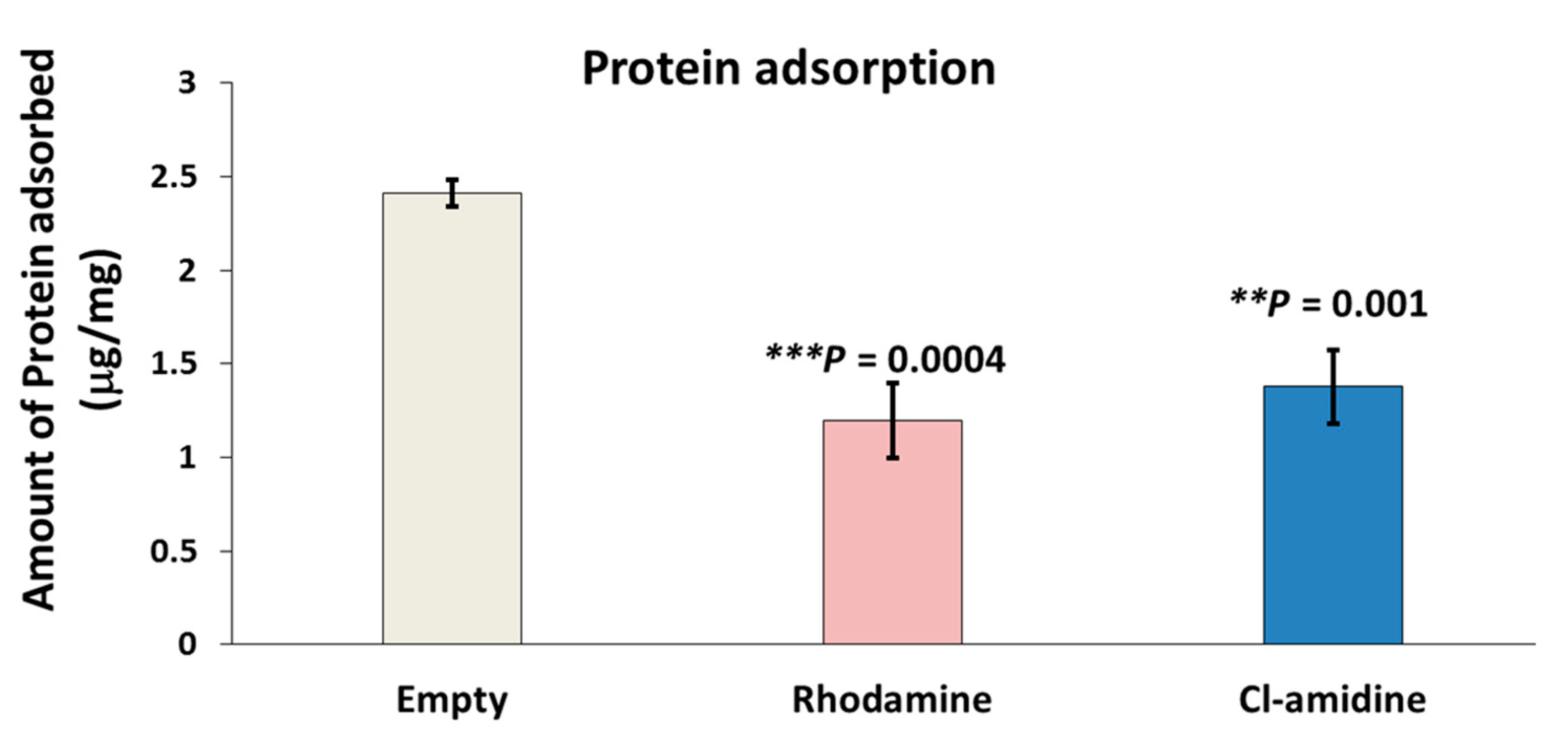
2.6. Protein Adsorption Assay (Bradford’s Assay)
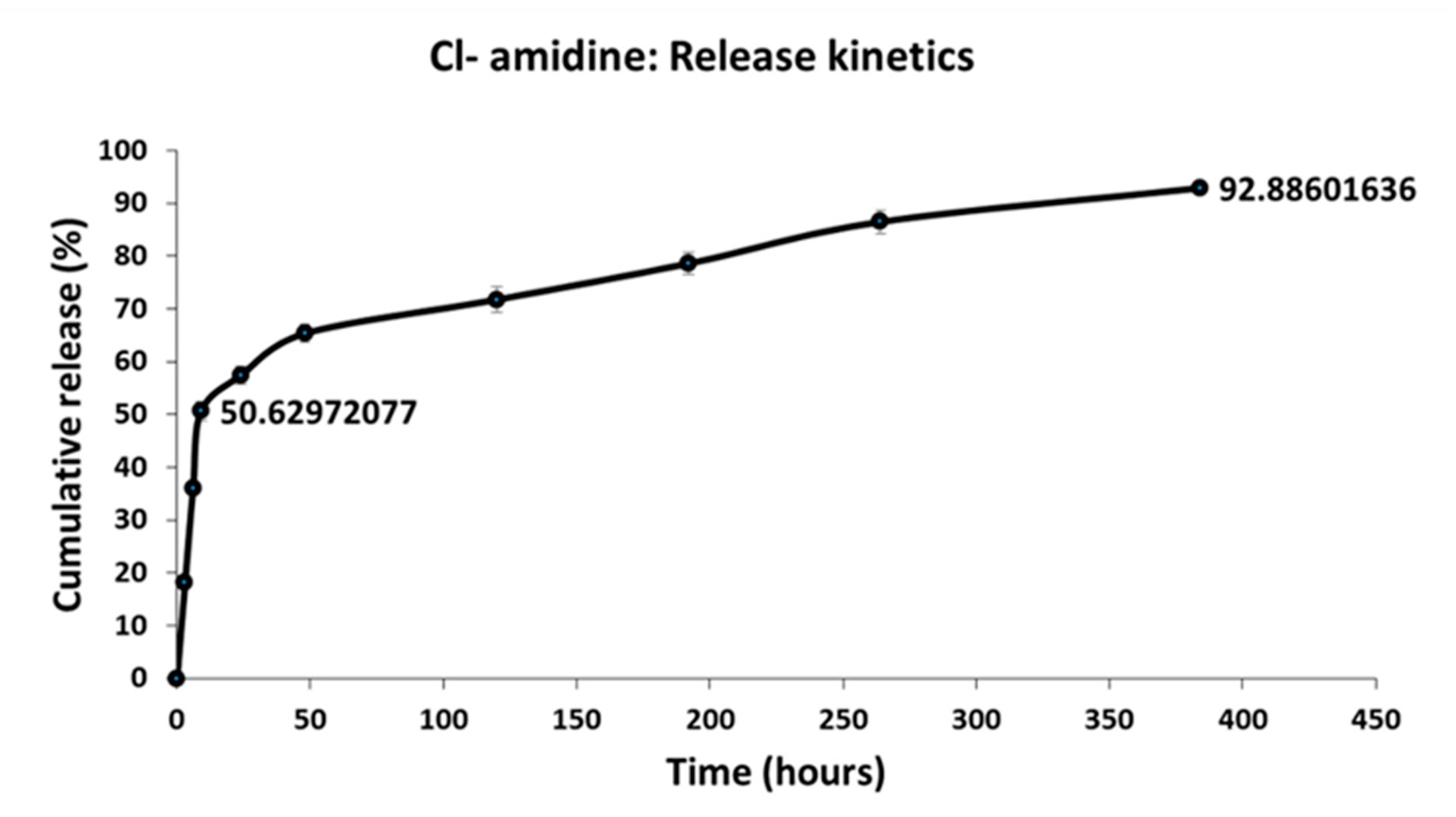
2.7. Cl-Amidine Encapsulation Efficiency and In Vitro Release Kinetics
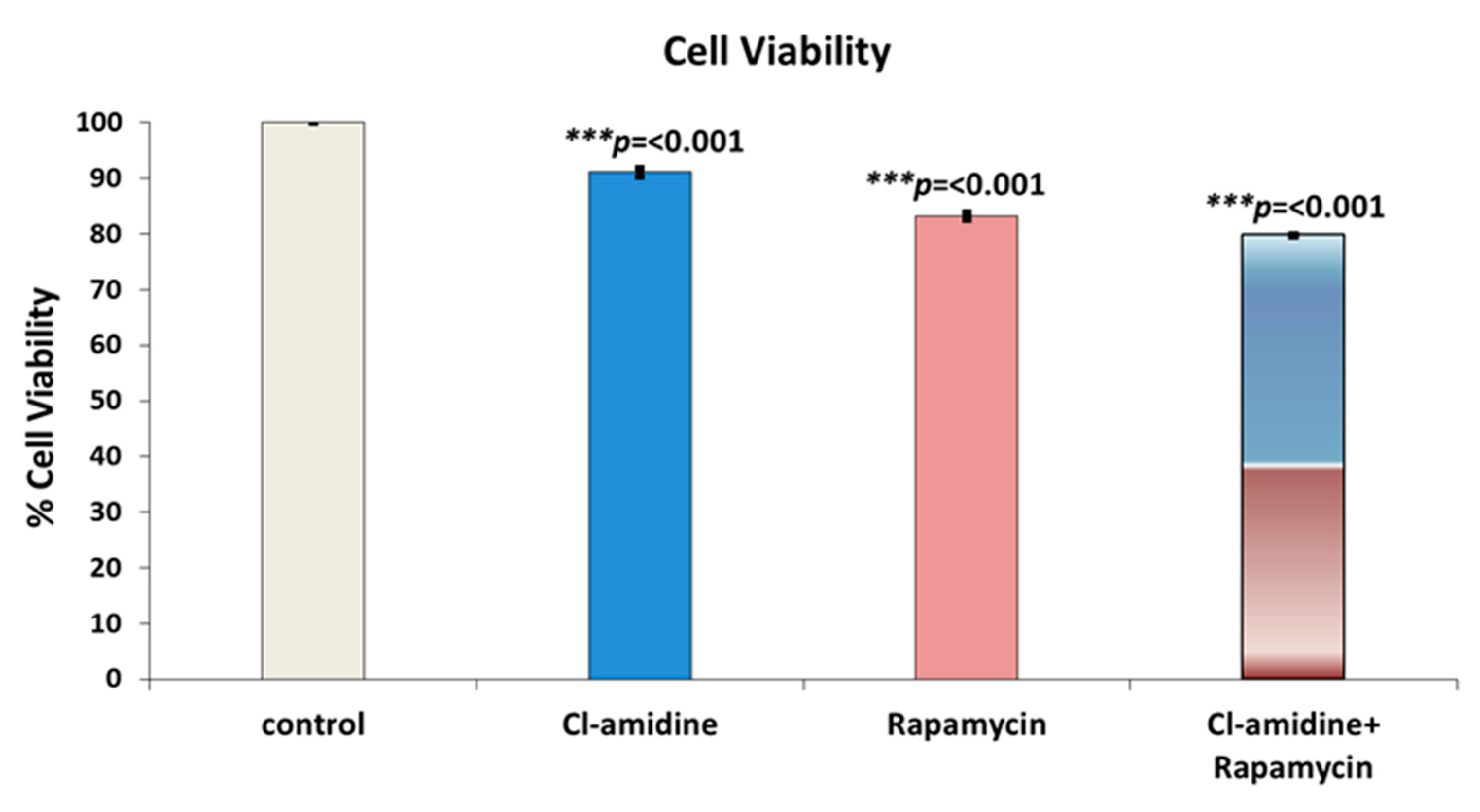
2.8. Cell Culture
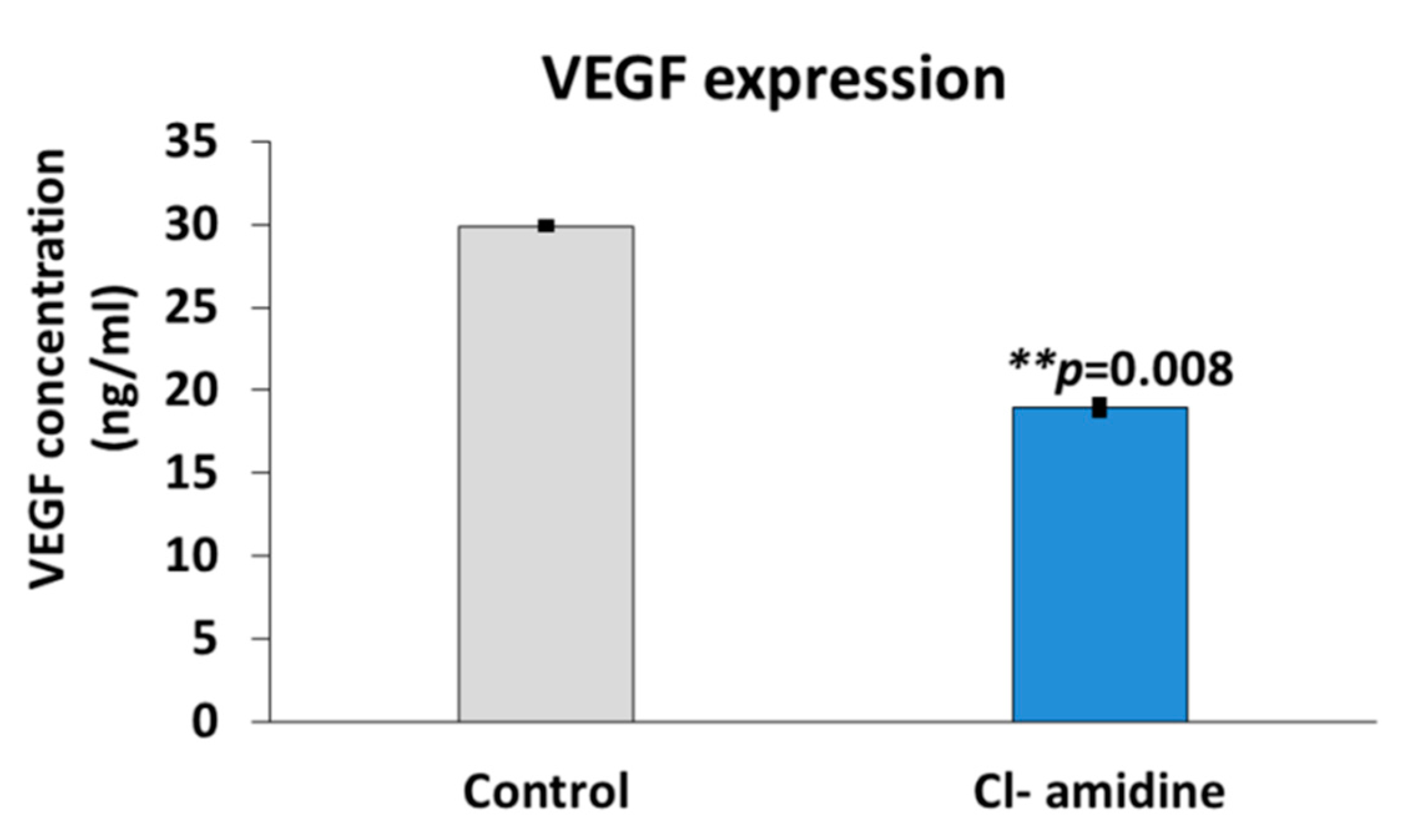
2.9. Vascular Endothelial Growth Factor Expression (VEGF)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Production of P(3HB) Microspheres with Encapsulated Rhodamine and Cl-Amidine
4.3. Characterization of P(3HB) Microspheres with and without Encapsulated Rhodamine or Cl-Amidine
4.3.1. Surface Morphology and Microstructure Characterization
4.3.2. Porosity
4.3.3. Determination of Residual PVA content
4.3.4. Determination of Surface Hydrophobicity
4.3.5. Protein Adsorption Studies Using Bradford’s Assay
4.4. In Vitro Drug Release Kinetics
4.5. Drug Quantification through Encapsulation Efficiency Calculation
4.6. Cell Culture
4.7. Effects on VEGF Expression
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vossenaar, E.; Zendman, A.; van Venrooij, W.; Pruijn, G. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays 2003, 25, 1106–1118. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylargininedeiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Gallagher, M.; Kholia, S.; Kosgodage, U.; Hristova, M.; Hardy, J.; Inal, J. PeptidylarginineDeiminases—Roles in Cancer and Neurodegeneration and Possible Avenues for Therapeutic Intervention via Modulation of Exosome and Microvesicle (EMV) Release? Int. J. Mol. Sci. 2017, 18, 1196. [Google Scholar] [CrossRef] [PubMed]
- Bicker, K.; Thompson, P. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 2013, 99, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A. Citrullination in Cancer. Cancer Res. 2019, 79, 1274–1284. [Google Scholar] [CrossRef]
- Lange, S.; Gögel, S.; Leung, K.; Vernay, B.; Nicholas, A.; Causey, C.; Thompson, P.; Greene, N.; Ferretti, P. Protein deiminases: New players in the developmentally regulated loss of neural regenerative ability. Dev. Biol. 2011, 355, 205–214. [Google Scholar] [CrossRef]
- Lange, S.; Rocha-Ferreira, E.; Thei, L.; Mawjee, P.; Bennett, K.; Thompson, P.; Subramanian, V.; Nicholas, A.; Peebles, D.; Hristova, M.; et al. Peptidylargininedeiminases: Novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J. Neurochem. 2014, 130, 555–562. [Google Scholar] [CrossRef]
- Kosgodage, U.; Uysal-Onganer, P.; MacLatchy, A.; Kraev, I.; Chatterton, N.; Nicholas, A.; Inal, J.; Lange, S. Peptidylarginine Deiminases Post-Translationally Deiminate Prohibitin and Modulate Extracellular Vesicle Release and MicroRNAs in Glioblastoma Multiforme. Int. J. Mol. Sci. 2018, 20, 103. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; MacLatchy, A.; Mahmoud, R.; Kraev, I.; Thompson, P.; Inal, J.; Lange, S. Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines. Int. J. Mol. Sci. 2020, 21, 1495. [Google Scholar] [CrossRef] [PubMed]
- Uysal-Onganer, P.; D’Alessio, S.; Mortoglou, M.; Kraev, I.; Lange, S. Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. Int. J. Mol. Sci. 2021, 22, 1396. [Google Scholar] [CrossRef] [PubMed]
- Witalison, E.; Thompson, P.; Hofseth, L. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef]
- Tilvawala, R.; Thompson, P. Peptidyl arginine deiminases: Detection and functional analysis of protein citrullination. Curr. Opin. Struct. Biol. 2019, 59, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Moscarello, M.; Wood, D.; Ackerley, C.; Boulias, C. Myelin in multiple sclerosis is developmentally immature. J. Clin. Investig. 1994, 94, 146–154. [Google Scholar] [CrossRef]
- Subramanian, V.; Nicholas, A.; Thompson, P.; Ferretti, P. Modulation of calcium-induced cell death in human neural stem cells by the novel peptidylarginine deiminase–AIF pathway. Biochim. Biophys. Acta 2014, 1843, 1162–1171. [Google Scholar]
- Nicholas, A. Dual immunofluorescence study of citrullinated proteins in Parkinson diseased substantia nigra. Neurosci. Lett. 2011, 495, 26–29. [Google Scholar] [CrossRef]
- Ishigami, A.; Masutomi, H.; Handa, S.; Nakamura, M.; Nakaya, S.; Uchida, Y.; Saito, Y.; Murayama, S.; Jang, B.; Jeon, Y.; et al. Mass spectrometric identification of citrullination sites and immunohistochemical detection of citrullinated glial fibrillary acidic protein in Alzheimer’s disease brains. J. Neurosci. Res. 2015, 93, 1664–1674. [Google Scholar] [CrossRef]
- Yang, L.; Tan, D.; Piao, H. Myelin Basic Protein Citrullination in Multiple Sclerosis: A Potential Therapeutic Target for the Pathology. Neurochem. Res. 2016, 41, 1845–1856. [Google Scholar] [CrossRef]
- Sancandi, M.; Uysal-Onganer, P.; Kraev, I.; Mercer, A.; Lange, S. Protein Deimination Signatures in Plasma and Plasma-EVs and Protein Deimination in the Brain Vasculature in a Rat Model of Pre-Motor Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2743. [Google Scholar] [CrossRef]
- Lange, S. Peptidylarginine deiminases and extracellular vesicles: Prospective drug targets and biomarkers in central nervous system diseases and repair. Neural Regen Res. 2021, 16, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Bradford, C.; Ramos, I.; Cross, A.; Haddock, G.; McQuaid, S.; Nicholas, A.; Woodroofe, M. Localisation of citrullinated proteins in normal appearing white matter and lesions in the central nervous system in multiplesclerosis. J. Neuroimmunol. 2014, 273, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Musse, A.; Li, Z.; Ackerley, C.; Bienzle, D.; Lei, H.; Poma, R.; Harauz, G.; Moscarello, M.; Mastronardi, F. Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system. Dis. Model. Mech. 2008, 1, 229–240. [Google Scholar] [CrossRef]
- Nicholas, A.; Sambandam, T.; Echols, J.; Barnum, S. Expression of citrullinated proteins in murine experimental autoimmune encephalomyelitis. J. Comp. Neurol. 2005, 486, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wasilewski, E.; Chakka, S.; Bello, A.; Moscarello, M.; Kotra, L. Novel Inhibitors of Protein Arginine Deiminase with Potential Activity in Multiple Sclerosis Animal Model. J. Med. Chem. 2013, 56, 1715–1722. [Google Scholar] [CrossRef]
- Muth, A.; Subramanian, V.; Beaumont, E.; Nagar, M.; Kerry, P.; McEwan, P.; Srinath, H.; Clancy, K.; Parelkar, S.; Thompson, P. Development of a Selective Inhibitor of Protein Arginine Deiminase 2. J. Med. Chem. 2017, 60, 3198–3211. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Ohsawa, T.; Hiratsuka, M.; Taguchi, H.; Kobayashi, S.; Saito, Y.; Murayama, S.; Asaga, H.; Toda, T.; Kimura, N.; et al. Abnormal accumulation of citrullinated proteins catalyzed by peptidylargininedeiminase in hippocampal extracts from patients with Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 120–128. [Google Scholar] [CrossRef]
- Jang, B.; Kim, E.; Choi, J.; Jin, J.; Kim, J.; Ishigami, A.; Maruyama, N.; Carp, R.; Kim, Y.; Choi, E. Accumulation of Citrullinated Proteins by Up-Regulated PeptidylarginineDeiminase 2 in Brains of Scrapie-Infected Mice. Am. J. Pathol. 2008, 173, 1129–1142. [Google Scholar] [CrossRef]
- Lange, S. PeptidylarginineDeiminases as Drug Targets in Neonatal Hypoxic–Ischemic Encephalopathy. Front. Neurol. 2016, 7, 22. [Google Scholar] [CrossRef]
- Luo, Y.; Knuckley, B.; Lee, Y.; Stallcup, M.; Thompson, P. A Fluoroacetamidine-Based Inactivator of Protein Arginine Deiminase 4: Design, Synthesis, and in Vitro and in Vivo Evaluation. J. Am. Chem. Soc. 2006, 128, 1092–1093. [Google Scholar] [CrossRef]
- Fetz, A.; Neeli, I.; Buddington, K.; Read, R.; Smeltzer, M.; Radic, M.; Bowlin, G. Localized Delivery of Cl-Amidine From Electrospun Polydioxanone Templates to Regulate Acute Neutrophil NETosis: A Preliminary Evaluation of the PAD4 Inhibitor for Tissue Engineering. Front. Pharmacol. 2018, 9, 289. [Google Scholar] [CrossRef]
- Mangat, P.; Wegner, N.; Venables, P.; Potempa, J. Bacterial and human peptidylargininedeiminases: Targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res. Ther. 2010, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Willis, V.; Gizinski, A.; Banda, N.; Causey, C.; Knuckley, B.; Cordova, K.; Luo, Y.; Levitt, B.; Glogowska, M.; Chandra, P.; et al. N-α-Benzoyl-N5-(2-Chloro-1-Iminoethyl)-l-Ornithine Amide, a Protein Arginine Deiminase Inhibitor, Reduces the Severity of Murine Collagen-Induced Arthritis. J. Immunol. 2011, 186, 4396–4404. [Google Scholar] [CrossRef]
- Kholia, S.; Jorfi, S.; Thompson, P.; Causey, C.; Nicholas, A.; Inal, J.; Lange, S. A novel role for peptidylarginine deiminases in microvesicle release reveals therapeutic potential of PAD inhibition in sensitizing prostate cancer cells to chemotherapy. J. Extracell Vesicles 2015, 4, 26192. [Google Scholar] [CrossRef] [PubMed]
- Kosgodage, U.; Trindade, R.; Thompson, P.; Inal, J.; Lange, S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Miao, L.; Xue, T.; Qin, H.; Mondal, S.; Thompson, P.; Coonrod, S.; Liu, X.; Zhang, X. Inhibiting PAD2 enhances the anti-tumor effect of docetaxel in tamoxifen-resistant breast cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 414. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Li, P.; Venters, B.; Zheng, S.; Thompson, P.; Pugh, B.; Wang, Y. Histone Arg Modifications and p53 Regulate the Expression of OKL38, a Mediator of Apoptosis. J. Biol. Chem. 2008, 283, 20060–20068. [Google Scholar] [CrossRef] [PubMed]
- Francis, L.; Meng, D.; Locke, I.; Knowles, J.; Mordan, N.; Salih, V.; Boccaccini, A.; Roy, I. Novel poly(3-hydroxybutyrate) composite films containing bioactive glass nanoparticles for wound healing applications. Polym. Int. 2016, 65, 661–674. [Google Scholar] [CrossRef]
- Sukan, A.; Roy, I.; Keshavarz, T. Dual production of biopolymers from bacteria. Carbohydr. Polym. 2015, 126, 47–51. [Google Scholar] [CrossRef]
- Nigmatullin, R.; Thomas, P.; Lukasiewicz, B.; Puthussery, H.; Roy, I. Polyhydroxyalkanoates, a family of natural polymers, and their applications in drug delivery. J. Chem. Technol. Biotechnol. 2015, 90, 1209–1221. [Google Scholar] [CrossRef]
- Di Mascolo, D.; Basnett, P.; Palange, A.; Francardi, M.; Roy, I.; Decuzzi, P. Tuning core hydrophobicity of spherical polymeric nanoconstructs for docetaxel delivery. Polym. Int. 2016, 65, 741–746. [Google Scholar] [CrossRef]
- Lizarraga-Valderrama, L.; Taylor, C.; Claeyssens, F.; Haycock, J.; Knowles, J.; Roy, I. Unidirectional neuronal cell growth and differentiation on aligned polyhydroxyalkanoate blend microfibres with varying diameters. J. Tissue Eng. Regen. Med. 2019, 13, 1581–1594. [Google Scholar] [CrossRef]
- Piarali, S.; Marlinghaus, L.; Viebahn, R.; Lewis, H.; Ryadnov, M.; Groll, J.; Salber, J.; Roy, I. Activated Polyhydroxyalkanoate Meshes Prevent Bacterial Adhesion and Biofilm Development in Regenerative Medicine Applications. Front. Bioeng. Biotechnol. 2020, 8, 442. [Google Scholar] [CrossRef] [PubMed]
- Basnett, P.; Matharu, R.; Taylor, C.; Illangakoon, U.; Dawson, J.; Kanczler, J.; Behbehani, M.; Humphrey, E.; Majid, Q.; Lukasiewicz, B.; et al. Harnessing Polyhydroxyalkanoates and Pressurized Gyration for Hard and Soft Tissue Engineering. ACS Appl. Mater. Interfaces 2021, 13, 32624–32639. [Google Scholar] [CrossRef]
- Hicklin, D.; Ellis, L. Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Li, X.; Gao, Y.; Li, J.; Zhang, K.; Han, J.; Li, W.; Hao, Q.; Zhang, W.; Wang, S.; Zeng, C.; et al. FOXP3 inhibits angiogenesis by downregulating VEGF in breast cancer. Cell Death Dis. 2018, 9, 744. [Google Scholar] [CrossRef]
- Karami, E.; Sabatier, J.; Behdani, M.; Irani, S.; Kazemi-Lomedasht, F. A nanobody-derived mimotope against VEGF inhibits cancer angiogenesis. J. Enzym. Inhib. Med. Chem. 2020, 35, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Mukherjee, A.; Saha, D.; Dash, J.; Chatterjee, T. Poly-l-Lysine inhibits VEGF and c-Myc mediated tumor-angiogenesis and induces apoptosis in 2D and 3D tumor microenvironment of both MDA-MB-231 and B16F10 induced mice model. Int. J. Biol. Macromol. 2021, 183, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.; Shi, X.; Hao, S.; Zhang, F.; Guo, Z.; Gao, Y.; Guo, H.; Liu, L. Oridonin inhibits tumor angiogenesis and induces vessel normalization in experimental colon cancer. J. Cancer 2021, 12, 3257–3264. [Google Scholar] [CrossRef] [PubMed]
- Sköld, M.; Kanje, M. Vascular Endothelial Growth Factor in Central Nervous System Injuries—A Vascular Growth Factor Getting Nervous? Curr. Neurovasc. Res. 2008, 5, 246–259. [Google Scholar] [CrossRef]
- Theis, V.; Theiss, C. VEGF—A Stimulus for Neuronal Development and Regeneration in the CNS and PNS. Curr. Protein Pept Sci. 2018, 19, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, D.; Kurganov, E.; Furube, E.; Morita, M.; Miyata, S. VEGF- and PDGF-dependent proliferation of oligodendrocyte progenitor cells in the medulla oblongata after LPC-induced focal demyelination. J. Neuroimmunol. 2019, 332, 176–186. [Google Scholar] [CrossRef]
- Butreddy, A.; Gaddam, R.; Kommineni, N.; Dudhipala, N.; Voshavar, C. PLGA/PLA-Based Long-Acting Injectable Depot Microspheres in Clinical Use: Production and Characterization Overview for Protein/Peptide Delivery. Int. J. Mol. Sci. 2021, 22, 8884. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; De Marco, I. Controlled-release antihistamines using supercritical antisolvent process. J. Supercrit Fluids 2021, 171, 105201. [Google Scholar] [CrossRef]
- Badens, E.; Masmoudi, Y.; Mouahid, A.; Crampon, C. Current situation and perspectives in drug formulation by using supercritical fluid technology. J. Supercrit. Fluids 2018, 134, 274–283. [Google Scholar] [CrossRef]
- He, W.; Suo, Q.; Jiang, Z.; Shan, A.; Hong, H. Precipitation of ephedrine by SEDS process using a specially designed prefilming atomizer. J. Supercrit. Fluids 2004, 31, 101–110. [Google Scholar] [CrossRef]
- Oliveira, J.; Priamo, W.; Dalmolin, I.; Boschetto, D.; Mezzomo, N.; Ferreira, S. Micronization processes by supercritical fluid technologies: A short review on process design (2008–2012). Acta Sci. Technol. 2013, 35, 695–709. [Google Scholar] [CrossRef][Green Version]
- Taki, S.; Badens, E.; Charbit, G. Controlled release system formed by supercritical anti-solvent coprecipitation of a herbicide and a biodegradable polymer. J. Supercrit. Fluids 2001, 21, 61–70. [Google Scholar] [CrossRef]
- Ge, J.; Jacobson, G.; Lobovkina, T.; Holmberg, K.; Zare, R. Sustained release of nucleic acids from polymeric nanoparticles using microemulsion precipitation in supercritical carbon dioxide. Chem. Commun. 2010, 46, 9034–9036. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Huff, R.; Shekunov, B. Drug Encapsulation Using Supercritical Fluid Extraction of Emulsions. J. Pharm. Sci. 2006, 95, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Nobes, G.; Maysinger, D.; Marchessault, R. Polyhydroxyalkanoates: Materials for Delivery Systems. Drug Deliv. 1998, 5, 167–177. [Google Scholar] [CrossRef]
- Bazzo, G.; Lemos-Senna, E.; Gonçalves, M.; Pires, A. Effect of preparation conditions on morphology, drug content and release profiles of poly(hydroxybutyrate) microparticles containing piroxicam. J. Braz. Chem. Soc. 2008, 19, 914–921. [Google Scholar] [CrossRef]
- Basnett, P.; Marcello, E.; Lukasiewicz, B.; Panchal, B.; Nigmatullin, R.; Knowles, J.; Roy, I. Biosynthesis and characterization of a novel, biocompatible medium chain length polyhydroxyalkanoate by Pseudomonas mendocina CH50 using coconut oil as the carbon source. J. Mater. Sci. Mater. Med. 2018, 29, 179. [Google Scholar] [CrossRef]
- Rai, R.; Boccaccini, A.; Knowles, J.; Mordon, N.; Salih, V.; Locke, I.; Moshrefi-Torbati, M.; Keshavarz, T.; Roy, I. The homopolymer poly(3-hydroxyoctanoate) as a matrix material for soft tissue engineering. J. Appl. Polym. Sci. 2011, 122, 3606–3617. [Google Scholar] [CrossRef]
- Rai, R.; Keshavarz, T.; Roether, J.; Boccaccini, A.; Roy, I. Medium chain length polyhydroxyalkanoates, promising new biomedical materials for the future. Mater. Sci. Eng. R Rep. 2011, 72, 29–47. [Google Scholar] [CrossRef]
- Sukan, A.; Roy, I.; Keshavarz, T. Agro-Industrial Waste Materials as Substrates for the Production of Poly(3-Hydroxybutyric Acid). J. Biomater. Nanobiotechnol. 2014, 5, 229–240. [Google Scholar] [CrossRef]
- Akaraonye, E.; Filip, J.; Safarikova, M.; Salih, V.; Keshavarz, T.; Knowles, J.; Roy, I. P(3HB) Based Magnetic Nanocomposites: Smart Materials for Bone Tissue Engineering. J. Nanomater. 2016, 3897592, 1–14. [Google Scholar] [CrossRef]
- Ching, K.; Andriotis, O.; Li, S.; Basnett, P.; Su, B.; Roy, I.; Tare, R.; Sengers, B.; Stolz, M. Nanofibrous poly(3-hydroxybutyrate)/poly(3-hydroxyoctanoate) scaffolds provide a functional microenvironment for cartilage repair. J. Biomater. Appl. 2016, 31, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Bagdadi, A.; Safari, M.; Dubey, P.; Basnett, P.; Sofokleous, P.; Humphrey, E.; Locke, I.; Edirisinghe, M.; Terracciano, C.; Boccaccini, A.; et al. Poly(3-hydroxyoctanoate), a promising new material for cardiac tissue engineering. J. Tissue Eng. Regen. Med. 2017, 12, e495–e512. [Google Scholar] [CrossRef]
- Rai, R.; Roether, J.; Knowles, J.; Mordan, N.; Salih, V.; Locke, I.; Gordge, M.; McCormick, A.; Mohn, D.; Stark, W.; et al. Highly elastomeric poly(3-hydroxyoctanoate) based natural polymer composite for enhanced keratinocyte regeneration. Int. J. Polym. Mater. 2016, 66, 326–335. [Google Scholar] [CrossRef]
- Haraźna, K.; Cichoń, E.; Skibiński, S.; Witko, T.; Solarz, D.; Kwiecień, I.; Marcello, E.; Zimowska, M.; Socha, R.; Szefer, E.; et al. Physicochemical and Biological Characterisation of Diclofenac Oligomeric Poly(3-hydroxyoctanoate) Hybrids as β-TCP Ceramics Modifiers for Bone Tissue Regeneration. Int. J. Mol. Sci. 2020, 21, 9452. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Yang, Z.; Lu, H. Strategies of polyhydroxyalkanoates modification for the medical application in neural regeneration/nerve tissue engineering. Adv. Biosci. Biotechnol. 2013, 4, 731–740. [Google Scholar] [CrossRef]
- Mouriño, V.; Cattalini, J.; Roether, J.; Dubey, P.; Roy, I.; Boccaccini, A. Composite polymer-bioceramic scaffolds with drug delivery capability for bone tissue engineering. Expert Opin. Drug Deliv. 2013, 10, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Basnett, P.; Ching, K.; Stolz, M.; Knowles, J.; Boccaccini, A.; Smith, C.; Locke, I.; Keshavarz, T.; Roy, I. Novel Poly(3-hydroxyoctanoate)/Poly(3-hydroxybutyrate) blends for medical applications. React. Funct. Polym. 2013, 73, 1340–1348. [Google Scholar] [CrossRef]
- Francis, L.; Meng, D.; Knowles, J.; Keshavarz, T.; Boccaccini, A.; Roy, I. Controlled Delivery of Gentamicin Using Poly(3-hydroxybutyrate) Microspheres. Int. J. Mol. Sci. 2011, 12, 4294–4314. [Google Scholar] [CrossRef]
- Fokong, S.; Theek, B.; Wu, Z.; Koczera, P.; Appold, L.; Jorge, S.; Resch-Genger, U.; van Zandvoort, M.; Storm, G.; Kiessling, F.; et al. Image-guided, targeted and triggered drug delivery to tumors using polymer-based microbubbles. J. Control. Release 2012, 163, 75–81. [Google Scholar] [CrossRef]
- Khansari, S.; Duzyer, S.; Sinha-Ray, S.; Hockenberger, A.; Yarin, A.; Pourdeyhimi, B. Two-Stage Desorption-Controlled Release of Fluorescent Dye and Vitamin from Solution-Blown and Electrospun Nanofiber Mats Containing Porogens. Mol. Pharmaceut. 2013, 10, 4509–4526. [Google Scholar] [CrossRef] [PubMed]
- Randriamahefa, S.; Renard, E.; Guérin, P.; Langlois, V. Fourier Transform Infrared Spectroscopy for Screening and Quantifying Production of PHAs by Pseudomonas Grown on Sodium Octanoate. Biomacromolecules 2003, 4, 1092–1097. [Google Scholar] [CrossRef]
- Maia, J.; Santana, M.; Ré, M. The effect of some processing conditions on the characteristics of biodegradable microspheres obtained by an emulsion solvent evaporation process. Braz. J. Chem. Eng. 2004, 21, 1–12. [Google Scholar] [CrossRef]
- Bolourtchian, N.; Karimi, K.; Aboofazeli, R. Preparation and characterization of ibuprofen microspheres. J. Microencapsul. 2005, 22, 529–538. [Google Scholar] [CrossRef]
- Martin, D.; Williams, S. Medical applications of poly-4-hydroxybutyrate: A strong flexible absorbable biomaterial. Biochem. Eng. J. 2003, 16, 97–105. [Google Scholar] [CrossRef]
- Govender, S.; Pillay, V.; Chetty, D.; Essack, S.; Dangor, C.; Govender, T. Optimisation and characterisation of bioadhesive controlled release tetracycline microspheres. Int. J. Pharm. 2005, 306, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Kakade, S.; Hassan, D. Formulation and in vitro/in vivo Evaluation of Novel Biodegradable Microspheres for Treatment of Hormone Responsive Cancers. Int. J. Pharm. Investig. 2020, 10, 184–191. [Google Scholar] [CrossRef]
- Kassab, A.; Xu, K.; Denkbas, E.; Dou, Y.; Zhao, S.; Piskin, E. Rifampicin carrying polyhydroxybutyrate microspheres as a potential chemoembolization agent. J. Biomater. Sci. Polym. Ed. 1997, 8, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials 2001, 22, 231–241. [Google Scholar] [CrossRef]
- Zielhuis, S.; Nijsen, J.; Figueiredo, R.; Feddes, B.; Vredenberg, A.; van het Schip, A.; Hennink, W. Surface characteristics of holmium-loaded poly(l-lactic acid) microspheres. Biomaterials 2005, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Boury, F.; Ivanova, T.; Panaίotov, I.; Proust, J.; Bois, A.; Richou, J. Dynamic Properties of Poly(DL-lactide) and Polyvinyl Alcohol Monolayers at the Air/Water and Dichloromethane/Water Interfaces. J. Colloid. Interface Sci. 1995, 169, 380–392. [Google Scholar] [CrossRef]
- Lee, S.C.; Oh, J.T.; Jang, M.H.; Chung, S.I. Quantitative analysis of polyvinyl alcohol on the surface of poly (D, L-lactide-co-glycolide) microparticles prepared by solvent evaporation method: Effect of particle size and PVA concentration. J. Control. Release 1999, 59, 123–132. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Panyam, J.; Prabha, S.; Labhasetwar, V. Residual polyvinyl alcohol associated with poly (D, L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J. Control. Release 2002, 82, 105–114. [Google Scholar] [CrossRef]
- Kemala, T.; Budianto, E.; Soegiyono, B. Preparation and characterization of microspheres based on blend of poly(lactic acid) and poly(ɛ-caprolactone) with poly(vinyl alcohol) as emulsifier. Arab. J. Chem. 2012, 5, 103–108. [Google Scholar] [CrossRef]
- Fang, L.; Singh, R.; Waxman, L.; Zhao, C. Model Protein Adsorption on Polymers Explained by Hansen Solubility Parameters. J. Pharm. Sci. 2019, 108, 187–192. [Google Scholar] [CrossRef]
- Ali, M.; Walboomers, X.; Jansen, J.; Yang, F. Influence of formulation parameters on encapsulation of doxycycline in PLGA microspheres prepared by double emulsion technique for the treatment of periodontitis. J. Drug. Deliv. Sci. Technol. 2019, 52, 263–271. [Google Scholar] [CrossRef]
- Şahin, A.; Eke, G.; Buyuksungur, A.; Hasirci, N.; Hasirci, V. Nuclear targeting peptide-modified, DOX-loaded, PHBV nanoparticles enhance drug efficacy by targeting to Saos-2 cell nuclear membranes. J. Biomater. Sci. Polym. Ed. 2018, 29, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ding, Y.; Rai, R.; Roether, J.; Schubert, D.; Boccaccini, A. Preparation and characterization of PHBV microsphere/45S5 bioactive glass composite scaffolds with vancomycin releasing function. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 41, 320–328. [Google Scholar] [CrossRef]
- Feng, S.; Lu, F.; Wang, Y.; Suo, J. Comparison of the degradation and release behaviors of poly(lactide-co-glycolide)-methoxypoly(ethylene glycol) microspheres prepared with single- and double-emulsion evaporation methods. J. Appl. Polym. Sci. 2015, 132, 41943. [Google Scholar] [CrossRef]
- Miladi, K.; Sfar, S.; Fessi, H.; Elaissari, A. Encapsulation of alendronate sodium by nanoprecipitation and double emulsion: From preparation to in vitro studies. Ind. Crops Prod. 2015, 72, 24–33. [Google Scholar] [CrossRef]
- Huerta-Angeles, G.; Brandejsová, M.; Nigmatullin, R.; Kopecká, K.; Vágnerová, H.; Šmejkalová, D.; Roy, I.; Velebný, V. Synthesis of graft copolymers based on hyaluronan and poly(3-hydroxyalkanoates). Carbohydr. Polym. 2017, 171, 220–228. [Google Scholar] [CrossRef]
- Lima, T.; Feitosa, R.; dos Santos-Silva, E.; dos Santos-Silva, A.; Siqueira, E.; Machado, P.; Cornélio, A.; do Egito, E.; Fernandes-Pedrosa, M.; Farias, K.; et al. Improving Encapsulation of Hydrophilic Chloroquine Diphosphate into Biodegradable Nanoparticles: A Promising Approach against Herpes Virus Simplex-1 Infection. Pharmaceutics 2018, 10, 255. [Google Scholar] [CrossRef]
- Klose, D.; Siepmann, F.; Elkharraz, K.; Siepmann, J. PLGA-based drug delivery systems: Importance of the type of drug and device geometry. Int. J. Pharm. 2008, 354, 95–103. [Google Scholar] [CrossRef]
- Rosca, I.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Hernández, J.; Villalba-Rodríguez, A.; Romero-Castillo, K.; Zavala-Yoe, R.; Bilal, M.; Ramirez-Mendoza, R.; Parra-Saldivar, R.; Iqbal, H. Poly-3-hydroxybutyrate-based constructs with novel characteristics for drug delivery and tissue engineering applications—A review. Polym. Eng. Sci. 2020, 60, 1760–1772. [Google Scholar] [CrossRef]
- Abe, H.; Doi, Y. Structural effects on enzymatic degradabilities for poly[(R)-3-hydroxybutyric acid] and its copolymers. Int. J. Biol. Macromol. 1999, 25, 185–192. [Google Scholar] [CrossRef]
- Burkersroda, F.; Schedl, L.; Göpferich, A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef]
- Lyu, S.; Untereker, D. Degradability of Polymers for Implantable Biomedical Devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef]
- Ulery, B.; Nair, L.; Laurencin, C. Biomedical applications of biodegradable polymers. J. Polym. Sci. B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef]
- Determan, A.; Trewyn, B.; Lin, V.; Nilsen-Hamilton, M.; Narasimhan, B. Encapsulation, stabilization, and release of BSA-FITC from polyanhydride microspheres. J. Control. Release 2004, 100, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M. Development of Drug Delivery Systems and Quality by Design. Recent Pat. Drug Deliv. Formul. 2015, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Vilos, C.; Velasquez, L. Therapeutic Strategies Based on Polymeric Microparticles. J. Biomed. Biotechnol. 2012, 2012, 672760. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Perrie, Y.; Coombes, A. Delivery of the antibiotic gentamicin sulphate from precipitation cast matrices of polycaprolactone. J. Control. Release 2006, 110, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Horii, F. Investigation of the structure of poly(vinyl alcohol)–iodine complex hydrogels prepared from the concentrated polymer solutions. Polymer 2008, 49, 785–791. [Google Scholar] [CrossRef]
- Fogh, J.; Fogh, J.; Orfeo, T. One Hundred and Twenty-Seven Cultured Human Tumor Cell Lines Producing Tumors in Nude Mice23. J. Natl. Cancer Inst. 1977, 59, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, H.; Liu, J.; Haley, K.; Treadway, J.; Larson, J.; Ge, N.; Peale, F.; Bruchez, M. Erratum: Corrigendum: Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat. Biotechnol. 2003, 21, 452. [Google Scholar] [CrossRef]
- Van de Loosdrecht, A.; Beelen, R.; Ossenkoppele, G.; Broekhoven, M.; Langenhuijsen, M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Drug | Drug Loading (mg) | P(3HB) Concentration (g/mL) | PVA Concentration (%w/v) | Stirrer Rate (rpm) |
|---|---|---|---|---|---|
| 1 | Cl-amidine | 2 | 0.013 | 0.5% | 300 |
| 2 | Cl-amidine | 2 | 0.013 | 1% | 300 |
| 3 | Cl-amidine | 2 | 0.0065 | 1% | 1200 |
| 3 | Rhodamine Control | 2 | 0.0065 | 1% | 1200 |
| 3 | None/Control | - | 0.0065 | 1% | 1200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, D.; Puthussery, H.; Basnett, P.; Knowles, J.C.; Lange, S.; Roy, I. Controlled Delivery of Pan-PAD-Inhibitor Cl-Amidine Using Poly(3-Hydroxybutyrate) Microspheres. Int. J. Mol. Sci. 2021, 22, 12852. https://doi.org/10.3390/ijms222312852
Ahmed D, Puthussery H, Basnett P, Knowles JC, Lange S, Roy I. Controlled Delivery of Pan-PAD-Inhibitor Cl-Amidine Using Poly(3-Hydroxybutyrate) Microspheres. International Journal of Molecular Sciences. 2021; 22(23):12852. https://doi.org/10.3390/ijms222312852
Chicago/Turabian StyleAhmed, Dina, Hima Puthussery, Pooja Basnett, Jonathan C. Knowles, Sigrun Lange, and Ipsita Roy. 2021. "Controlled Delivery of Pan-PAD-Inhibitor Cl-Amidine Using Poly(3-Hydroxybutyrate) Microspheres" International Journal of Molecular Sciences 22, no. 23: 12852. https://doi.org/10.3390/ijms222312852
APA StyleAhmed, D., Puthussery, H., Basnett, P., Knowles, J. C., Lange, S., & Roy, I. (2021). Controlled Delivery of Pan-PAD-Inhibitor Cl-Amidine Using Poly(3-Hydroxybutyrate) Microspheres. International Journal of Molecular Sciences, 22(23), 12852. https://doi.org/10.3390/ijms222312852