
Evaluation of Deep Neural Network ProSPr for Accurate Protein Distance Predictions on CASP14 Targets

 ,
,
Abstract
:1. Introduction
2. Evaluation and Results
3. Methods
3.1. ProSPr Overview
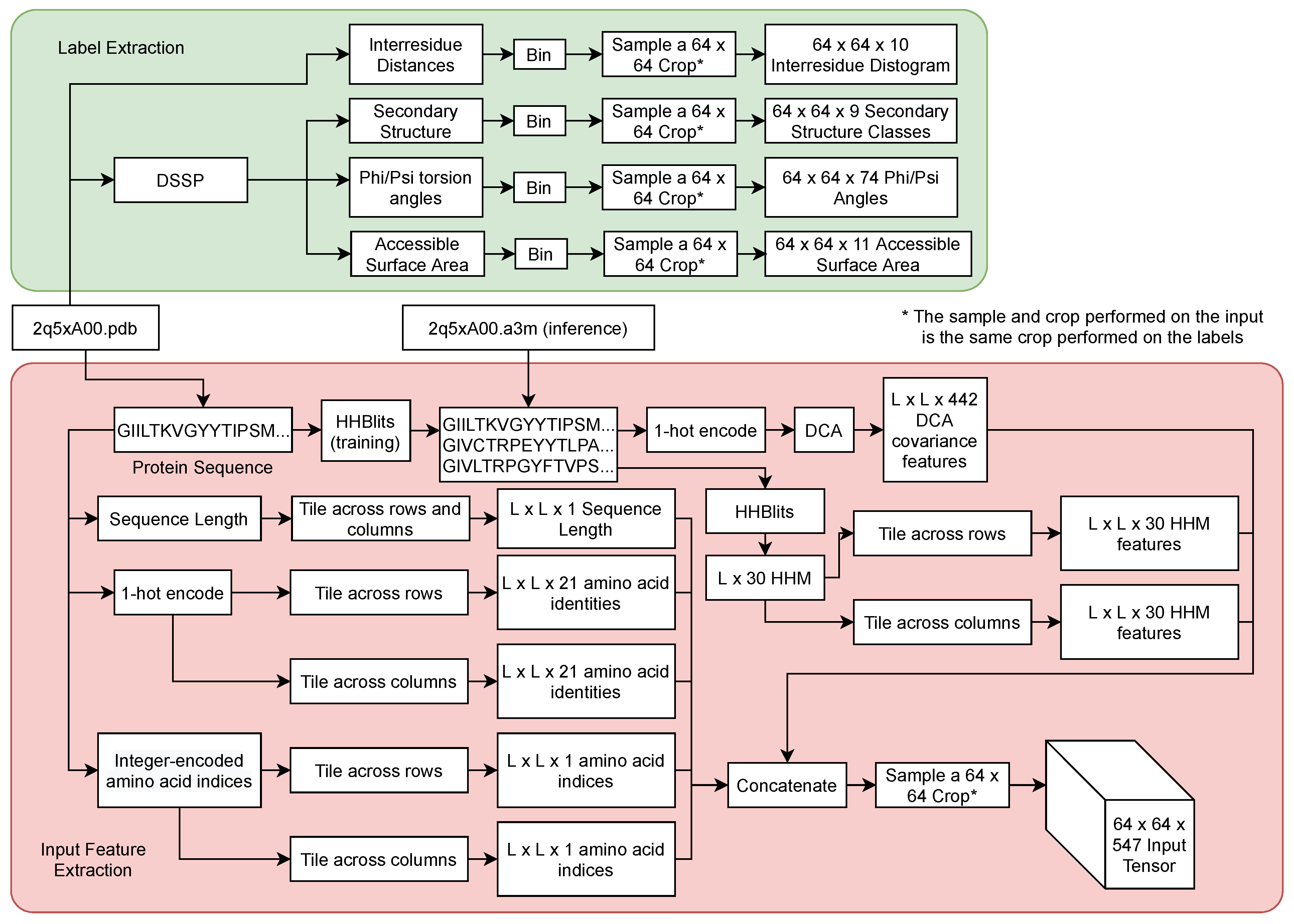
3.2. Input Features
3.3. Training Data
3.4. Training Strategy
3.5. Inference
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Della Corte, D.; van Beek, H.L.; Syberg, F.; Schallmey, M.; Tobola, F.; Cormann, K.U.; Schlicker, C.; Baumann, P.T.; Krumbach, K.; Sokolowsky, S. Engineering and application of a biosensor with focused ligand specificity. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Morris, C.J.; Corte, D.D. Using molecular docking and molecular dynamics to investigate protein-ligand interactions. Mod. Phys. Lett. B 2021, 35, 2130002. [Google Scholar] [CrossRef]
- Coates, T.L.; Young, N.; Jarrett, A.J.; Morris, C.J.; Moody, J.D.; Corte, D.D. Current computational methods for enzyme design. Mod. Phys. Lett. B 2021, 35, 2150155. [Google Scholar] [CrossRef]
- Möckel, C.; Kubiak, J.; Schillinger, O.; Kühnemuth, R.; Della Corte, D.; Schröder, G.F.; Willbold, D.; Strodel, B.; Seidel, C.A.; Neudecker, P. Integrated NMR, fluorescence, and molecular dynamics benchmark study of protein mechanics and hydrodynamics. J. Phys. Chem. B 2018, 123, 1453–1480. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikovet, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Horovitz, A. Extending the new generation of structure predictors to account for dynamics and allostery. J. Mol. Biol. 2021, 433, 167007. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.; Bridgland, A. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.; Bridgland, A. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Billings, W.M.; Hedelius, B.; Millecam, T.; Wingate, D.; Della Corte, D. ProSPr: Democratized implementation of alphafold protein distance prediction network. BioRxiv 2019, 830273. [Google Scholar] [CrossRef] [Green Version]
- CASP. CASP14 Abstracts. Available online: https://predictioncenter.org/casp14/doc/CASP14_Abstracts.pdf (accessed on 24 November 2021).
- Billings, W.M.; Morris, C.J.; Della Corte, D. The whole is greater than its parts: Ensembling improves protein contact prediction. Sci. Rep. 2021, 11, 1–7. [Google Scholar]
- Xu, J.; Wang, S. Analysis of distance-based protein structure prediction by deep learning in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1069–1081. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Terashi, G.; Kagaya, Y.; Subramaniya, S.R.M.V.; Christoffer, C.; Kihara, D. Analyzing effect of quadruple multiple sequence alignments on deep learning based protein inter-residue distance prediction. Sci. Rep. 2021, 11, 1–13. [Google Scholar]
- Li, Y.; Zhang, C.; Bell, E.W.; Yu, D.J.; Zhang, Y. Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1082–1091. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, J.; Guo, Z.; Wu, T.; Hou, J.; Cheng, J. Protein model accuracy estimation empowered by deep learning and inter-residue distance prediction in CASP14. Sci. Rep. 2021, 11, 1–12. [Google Scholar]
- Shrestha, R.; Fajardo, E.; Gil, N.; Fidelis, K.; Kryshtafovych, A.; Monastyrskyy, B.; Fiser, A. Assessing the accuracy of contact predictions in CASP13. Proteins 2019, 87, 1058–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, S.; Oruc, T.; Mead, L.; Rehman, M.F.; Thomas, C.M.; Butterworth, S.; Winn, P.J. DeepCDpred: Inter-residue distance and contact prediction for improved prediction of protein structure. PLoS ONE 2019, 14, e0205214. [Google Scholar] [CrossRef] [Green Version]
- Torrisi, M.; Pollastri, G. Protein structure annotations. In Essentials of Bioinformatics; Springer: Cham, Switzerland, 2019; Volume I, pp. 201–234. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Santurkar, S.; Tsipras, D.; Ilyas, A.; Mądry, A. How does batch normalization help optimization? In Proceedings of the 32nd International Conference on Neural Information Processing Systems, Montreal, QC, Canada, 3–8 December 2018; pp. 2488–2498. [Google Scholar]
- Clevert, D.-A.; Unterthiner, T.; Hochreiter, S. Fast and accurate deep network learning by exponential linear units (elus). arXiv 2015, arXiv:1511.07289. [Google Scholar]
- Yu, F.; Koltun, V. Multi-scale context aggregation by dilated convolutions. arXiv 2015, arXiv:1511.07122. [Google Scholar]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, M.; Wiuf, C. The CATH database. Hum. Genom. 2010, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Moult, J.; Billings, W.M.; Della Corte, D.; Fidelis, K.; Kwon, S.; Olechnovič, K.; Seok, C.; Venclovas, Č.; Won, J. Modeling SARS-CoV2 proteins in the CASP-commons experiment. Proteins Struct. Funct. Bioinform. 2021, 89, 1987–1996. [Google Scholar] [CrossRef]
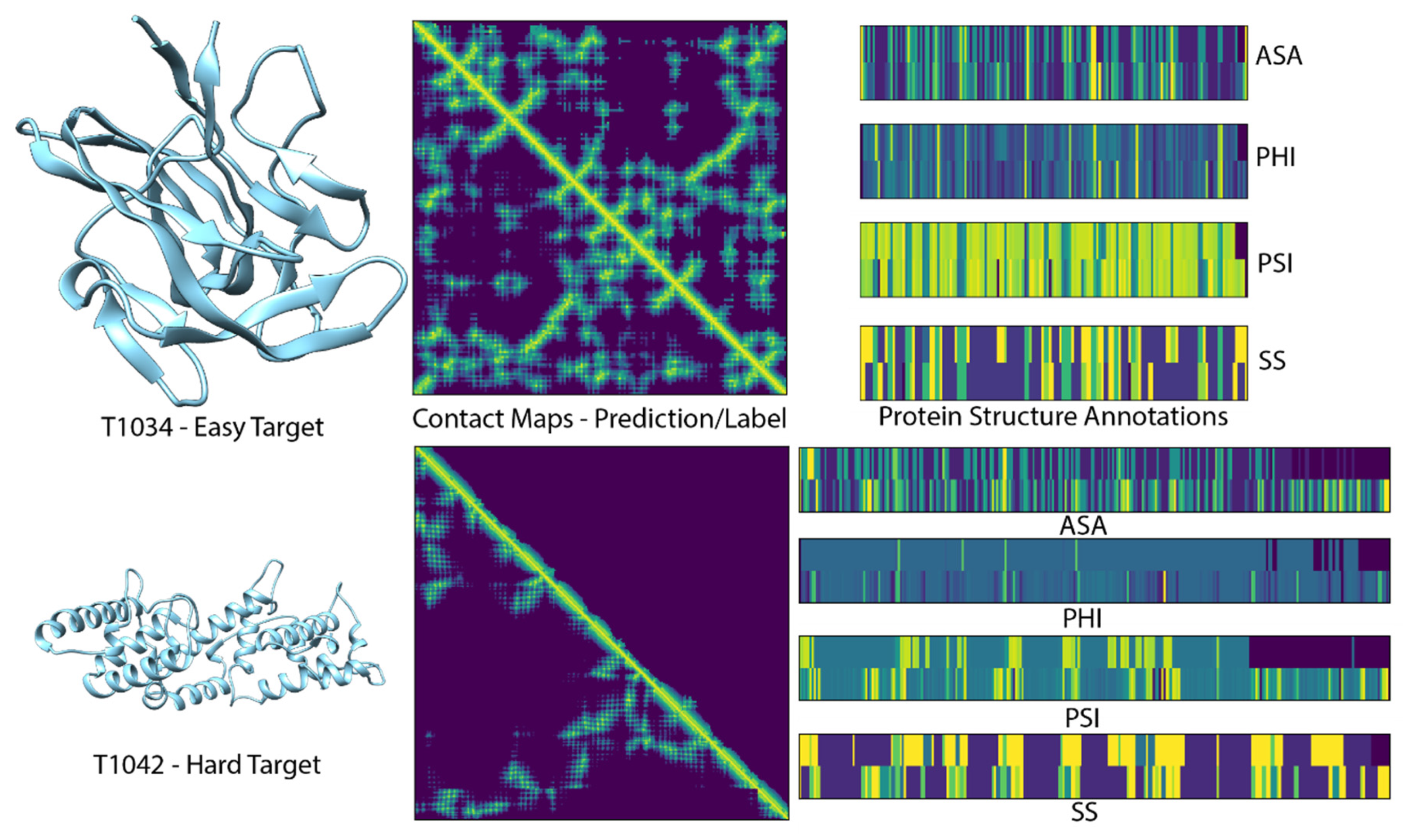



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ProSPr Model | Contact Accuracy (%) | |||
|---|---|---|---|---|
| Short | Mid | Long | Average | |
| A | 81.09% | 69.52% | 41.63% | 64.08% |
| B | 81.15% | 69.29% | 42.41% | 64.28% |
| C | 81.94% | 69.97% | 43.59% | 65.17% |
| Ensemble | 82.08% | 70.55% | 44.04% | 65.56% |
| Target | Contact Accuracy | ||
|---|---|---|---|
| Short | Mid | Long | |
| T1045s2 | 0.833 | 0.924 | 0.694 |
| T1046s1 | 1.000 | 1.000 | 0.536 |
| T1046s2 | 0.892 | 0.574 | 0.303 |
| T1047s1 | 0.907 | 0.985 | 0.639 |
| T1047s2 | 1.000 | 0.983 | 0.852 |
| T1060s2 | 0.857 | 0.575 | 0.282 |
| T1060s3 | 0.976 | 0.955 | 0.793 |
| T1065s1 | 1.000 | 0.973 | 0.518 |
| T1065s2 | 1.000 | 1.000 | 0.870 |
| T1024 | 1.000 | 1.000 | 0.809 |
| T1026 | 0.750 | 0.425 | 0.494 |
| T1027 | 0.485 | 0.278 | 0.054 |
| T1029 | 0.891 | 0.818 | 0.220 |
| T1030 | 0.804 | 0.792 | 0.333 |
| T1031 | 0.686 | 0.457 | 0.105 |
| T1032 | 0.889 | 0.851 | 0.580 |
| T1033 | 0.750 | 0.316 | 0.216 |
| T1034 | 0.988 | 0.874 | 0.885 |
| T1035 | 0.412 | 0.080 | 0.000 |
| T1037 | 0.690 | 0.455 | 0.030 |
| T1038 | 0.720 | 0.538 | 0.407 |
| T1039 | 0.269 | 0.000 | 0.007 |
| T1040 | 0.318 | 0.222 | 0.027 |
| T1041 | 0.644 | 0.357 | 0.021 |
| T1042 | 0.487 | 0.441 | 0.058 |
| T1043 | 0.431 | 0.216 | 0.014 |
| T1049 | 1.000 | 0.939 | 0.440 |
| T1050 | 0.964 | 0.821 | 0.705 |
| T1052 | 0.728 | 0.600 | 0.417 |
| T1053 | 0.796 | 0.521 | 0.093 |
| T1054 | 1.000 | 1.000 | 0.710 |
| T1055 | 0.932 | 0.860 | 0.200 |
| T1056 | 0.823 | 0.829 | 0.661 |
| T1057 | 1.000 | 0.987 | 0.815 |
| T1058 | 0.821 | 0.678 | 0.678 |
| T1061 | 0.807 | 0.687 | 0.511 |
| T1064 | 0.615 | 0.500 | 0.094 |
| T1067 | 0.865 | 0.824 | 0.466 |
| T1068 | 0.926 | 0.813 | 0.204 |
| T1070 | 0.941 | 0.707 | 0.579 |
| T1073 | 1.000 | 1.000 | 1.000 |
| T1074 | 0.845 | 0.700 | 0.328 |
| T1076 | 0.970 | 0.947 | 0.911 |
| T1078 | 0.984 | 0.892 | 0.587 |
| T1079 | 0.956 | 0.964 | 0.739 |
| T1082 | 0.615 | 0.636 | 0.164 |
| T1083 | 0.909 | 0.783 | 0.909 |
| T1084 | 1.000 | 1.000 | 1.000 |
| T1087 | 1.000 | 0.810 | 0.714 |
| T1088 | 0.954 | 1.000 | 0.778 |
| T1089 | 0.972 | 0.813 | 0.624 |
| T1090 | 0.977 | 0.870 | 0.399 |
| T1091 | 0.832 | 0.571 | 0.071 |
| T1092 | 0.704 | 0.782 | 0.382 |
| T1093 | 0.673 | 0.519 | 0.109 |
| T1094 | 0.649 | 0.580 | 0.144 |
| T1095 | 0.722 | 0.711 | 0.448 |
| T1096 | 0.766 | 0.421 | 0.098 |
| T1099 | 0.800 | 0.375 | 0.101 |
| T1100 | 0.883 | 0.820 | 0.258 |
| T1101 | 0.960 | 0.988 | 0.783 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stern, J.; Hedelius, B.; Fisher, O.; Billings, W.M.; Della Corte, D. Evaluation of Deep Neural Network ProSPr for Accurate Protein Distance Predictions on CASP14 Targets. Int. J. Mol. Sci. 2021, 22, 12835. https://doi.org/10.3390/ijms222312835
Stern J, Hedelius B, Fisher O, Billings WM, Della Corte D. Evaluation of Deep Neural Network ProSPr for Accurate Protein Distance Predictions on CASP14 Targets. International Journal of Molecular Sciences. 2021; 22(23):12835. https://doi.org/10.3390/ijms222312835
Chicago/Turabian StyleStern, Jacob, Bryce Hedelius, Olivia Fisher, Wendy M. Billings, and Dennis Della Corte. 2021. "Evaluation of Deep Neural Network ProSPr for Accurate Protein Distance Predictions on CASP14 Targets" International Journal of Molecular Sciences 22, no. 23: 12835. https://doi.org/10.3390/ijms222312835
APA StyleStern, J., Hedelius, B., Fisher, O., Billings, W. M., & Della Corte, D. (2021). Evaluation of Deep Neural Network ProSPr for Accurate Protein Distance Predictions on CASP14 Targets. International Journal of Molecular Sciences, 22(23), 12835. https://doi.org/10.3390/ijms222312835