
MemDis: Predicting Disordered Regions in Transmembrane Proteins


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
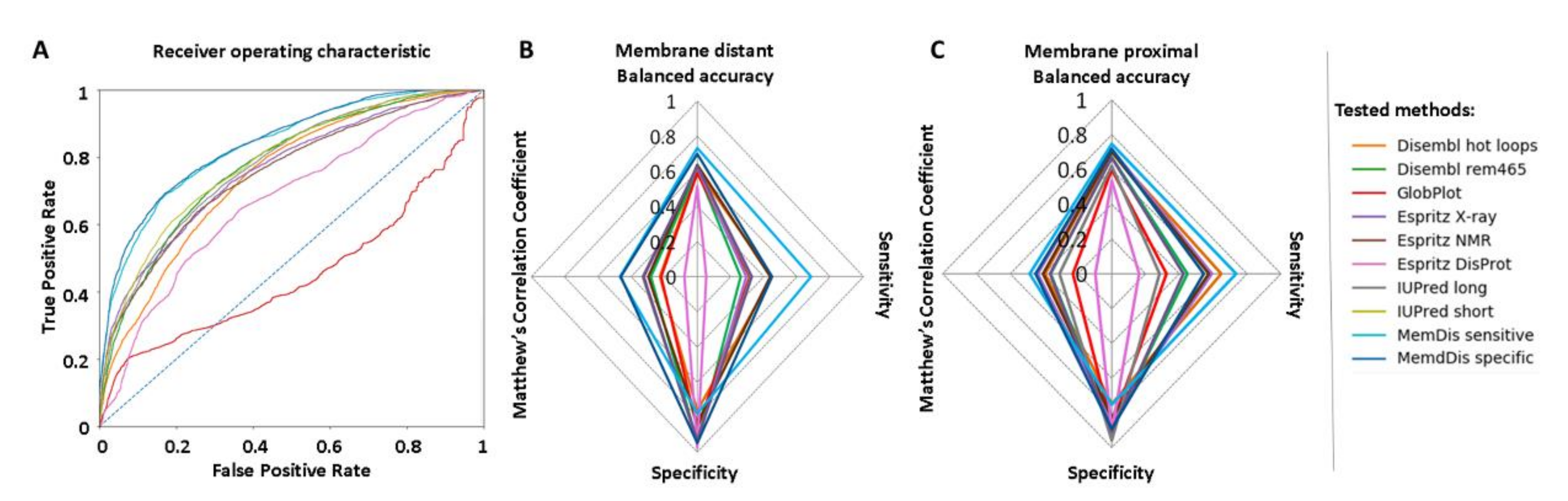
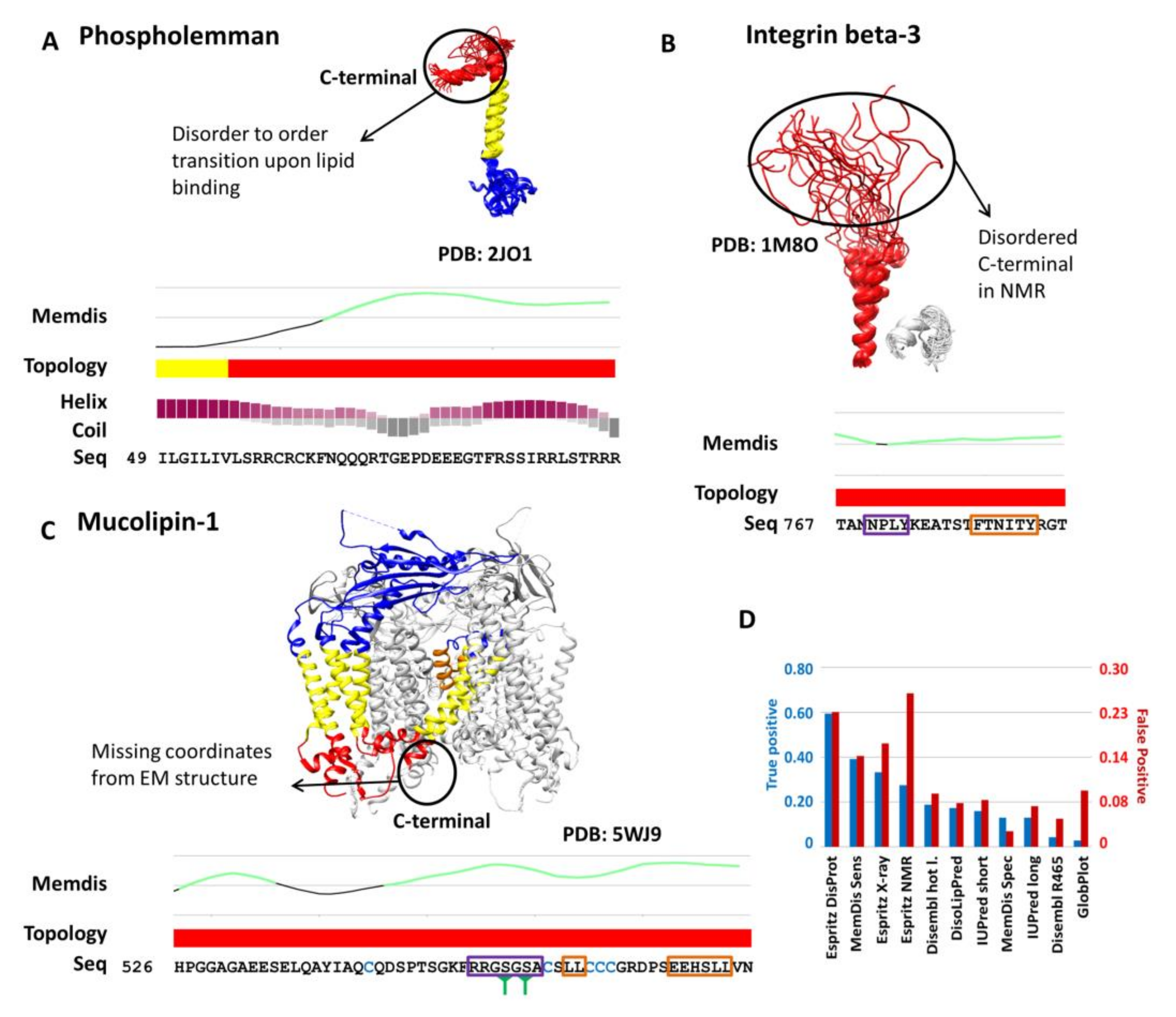
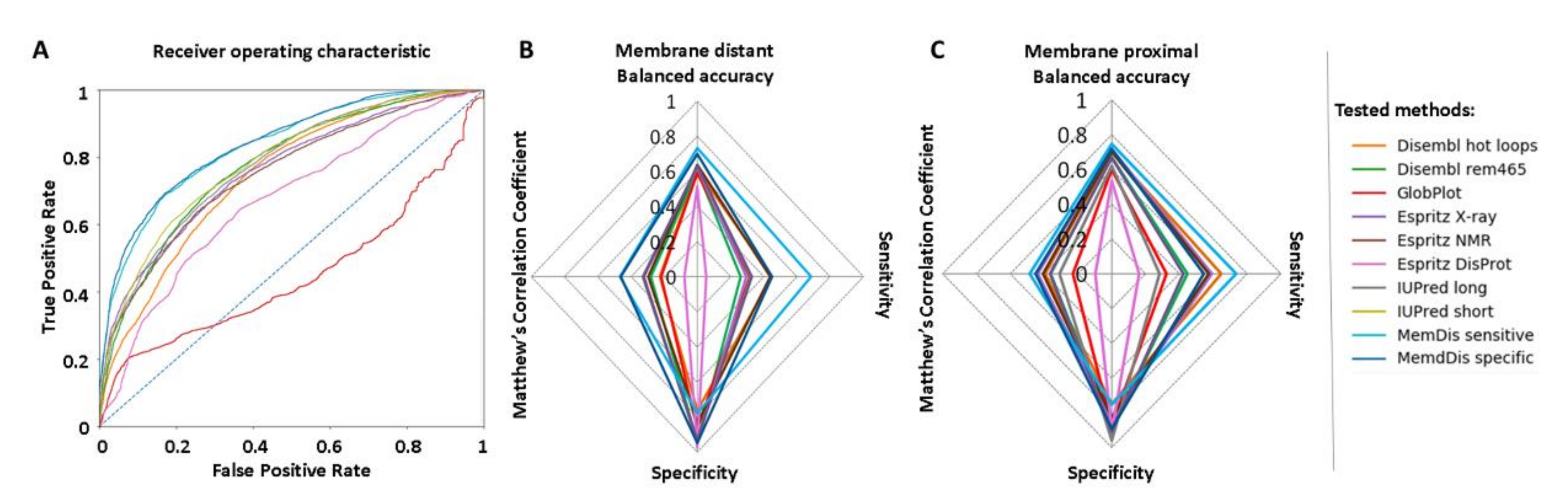
2. Results
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobson, L.; Reményi, I.; Tusnády, G.E. The human transmembrane proteome. Biol. Direct 2015, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaergaard, M.; Kragelund, B.B. Functions of intrinsic disorder in transmembrane proteins. Cell. Mol. Life Sci. 2017, 74, 3205–3224. [Google Scholar] [CrossRef] [PubMed]
- Tusnády, G.E.; Dobson, L.; Tompa, P. Disordered regions in transmembrane proteins. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2839–2848. [Google Scholar] [CrossRef] [Green Version]
- Reichmann, D.; Jakob, U. The roles of conditional disorder in redox proteins. Curr. Opin. Struct. Biol. 2013, 23, 436–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdős, G.; Pajkos, M.; Dosztányi, Z. IUPred3: Prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021, 49, W297–W303. [Google Scholar] [CrossRef]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2011, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Necci, M.; Piovesan, D.; Tosatto, S.C. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef]
- Cheung, J.Y.; Zhang, X.-Q.; Song, J.; Gao, E.; Rabinowitz, J.E.; Chan, T.O.; Wang, J. Phospholemman: A Novel Cardiac Stress Protein. Clin. Transl. Sci. 2010, 3, 189–196. [Google Scholar] [CrossRef]
- Teriete, P.; Franzin, C.M.; Choi, J.; Marassi, F.M. Structure of the Na, K-ATPase Regulatory Protein FXYD1 in Micelles. Biochemistry 2007, 46, 6774–6783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Walsh, I.; Minervini, G.; Tosatto, S.C.E. FELLS: Fast estimator of latent local structure. Bioinformatics 2017, 33, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, F.; Mészáros, B.; Salladini, E.; Hatos, A.; Pancsa, R.; Chemes, B.L.; Pajkos, M.; Lazar, T.; Pena-Diaz, S.; Santos, J.; et al. DisProt in 2022: Improved quality and accessibility of protein intrinsic disorder. Nucleic Acids Res. 2022. [Google Scholar]
- Mészáros, B.; Sámano-Sánchez, H.; Alvarado-Valverde, J.; Čalyševa, J.; Martínez-Pérez, E.; Alves, R.; Shields, D.C.; Kumar, M.; Rippmann, F.; Chemes, L.B.; et al. Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. Sci. Signal. 2021, 14, eabd0334. [Google Scholar] [CrossRef]
- Schmiege, P.; Fine, M.; Blobel, G.; Li, X. Human TRPML1 channel structures in open and closed conformations. Nature 2017, 550, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Vergarajauregui, S.; Puertollano, R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 2006, 7, 337–353. [Google Scholar] [CrossRef] [Green Version]
- Vergarajauregui, S.; Oberdick, R.; Kiselyov, K.; Puertollano, R. Mucolipin 1 channel activity is regulated by protein kinase A-mediated phosphorylation. Biochem. J. 2008, 410, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Mészáros, B.; Sámano-Sánchez, H.; Zeke, A.; Dobson, L.; Lazar, T.; Örd, M.; Nagpal, A.; et al. The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res. 2021, gkab975. [Google Scholar] [CrossRef]
- Csizmadia, G.; Erdős, G.; Tordai, H.; Padányi, R.; Tosatto, S.; Dosztányi, Z.; Hegedűs, T. The MemMoRF database for recognizing disordered protein regions interacting with cellular membranes. Nucleic Acids Res. 2020, 49, D355–D360. [Google Scholar] [CrossRef]
- Katuwawala, A.; Zhao, B.; Kurgan, L. DisoLipPred: Accurate prediction of disordered lipid binding residues in protein sequences with deep recurrent networks and transfer learning. Bioinformatics 2021, 93, btab640. [Google Scholar] [CrossRef]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Mičetić, I.; Camilloni, C.; Davey, N.; Dosztányi, Z.; Mészáros, B.; Monzon, A.M.; et al. MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018, 46, D471–D476. [Google Scholar] [CrossRef]
- Dobson, L.; Reményi, I.; Tusnády, G.E. CCTOP: A consensus constrained topology prediction web server. Nucleic Acids Res. 2015, 43, W408–W412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Jarnot, P.; Ziemska-Legięcka, J.; Dobson, L.; Merski, M.; Mier, P.; Andrade-Navarro, M.A.; Hancock, J.M.; Dosztányi, Z.; Paladin, L.; Necci, M.; et al. PlaToLoCo: The first web meta-server for visualization and annotation of low complexity regions in proteins. Nucleic Acids Res. 2020, 48, W77–W84. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobson, L.; Tusnády, G.E. MemDis: Predicting Disordered Regions in Transmembrane Proteins. Int. J. Mol. Sci. 2021, 22, 12270. https://doi.org/10.3390/ijms222212270
Dobson L, Tusnády GE. MemDis: Predicting Disordered Regions in Transmembrane Proteins. International Journal of Molecular Sciences. 2021; 22(22):12270. https://doi.org/10.3390/ijms222212270
Chicago/Turabian StyleDobson, Laszlo, and Gábor E. Tusnády. 2021. "MemDis: Predicting Disordered Regions in Transmembrane Proteins" International Journal of Molecular Sciences 22, no. 22: 12270. https://doi.org/10.3390/ijms222212270
APA StyleDobson, L., & Tusnády, G. E. (2021). MemDis: Predicting Disordered Regions in Transmembrane Proteins. International Journal of Molecular Sciences, 22(22), 12270. https://doi.org/10.3390/ijms222212270