
Proteomic Changes of Activated Hepatic Stellate Cells

 ,
,  ,
,  , , , and
, , , and 
Abstract
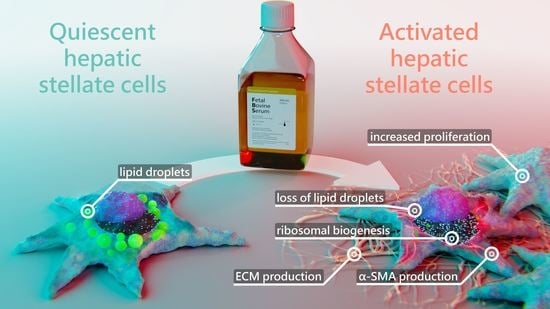
1. Introduction
2. Results
2.1. Hepatic Stellate Cells Are Activated by Fetal Bovine Serum
2.2. Serum Activated Hepatic Stellate Cells Show a Phenotype Characteristic of Activated Hepatic Stellate Cells
2.3. Proteomic Analysis of Serum Activated Hepatic Stellate Cells Reveals Changes of Several Key Cellular Pathways
2.3.1. Serum Activated Hepatic Stellate Cells Show Increased Ribosome Biogenesis, Cell Cycle, Cell Migration and Oxidative Stress Related Proteins
2.3.2. Activated Hepatic Stellate Cells Show a Decrease in Fatty Acid and Cholesterol Biosynthesis
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Proliferation Assay
4.3. Western Blotting Analysis
4.4. Proteomic Analysis
4.5. Proteomic Data Analysis
4.6. Migration Gap Closure Assay
4.7. Migration Transwell Assay
4.8. Lipid Droplet Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Kisseleva, T.; Brenner, D. Molecular and Cellular Mechanisms of Liver Fibrosis and Its Regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Bataller, R.; Gao, B. Liver Fibrosis in Alcoholic Liver Disease. Semin. Liver Dis. 2015, 35, 146–156. [Google Scholar] [CrossRef]
- Su, T.-H.; Kao, J.-H.; Liu, C.-J. Molecular Mechanism and Treatment of Viral Hepatitis-Related Liver Fibrosis. Int. J. Mol. Sci. 2014, 15, 10578–10604. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Zoubek, M.E.; Trautwein, C.; Strnad, P. Reversal of Liver Fibrosis: From Fiction to Reality. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 129–141. [Google Scholar] [CrossRef]
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the Background of Liver Fibrosis: Implications for the Management of Hepatocellular Cancer. World J. Gastroenterol. 2018, 24, 4436–4447. [Google Scholar] [CrossRef]
- Altamirano-Barrera, A.; Barranco-Fragoso, B.; Méndez-Sánchez, N. Management Strategies for Liver Fibrosis. Ann. Hepatol. 2017, 16, 48–56. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Haaker, M.W.; Vaandrager, A.B.; Helms, J.B. Retinoids in Health and Disease: A Role for Hepatic Stellate Cells in Affecting Retinoid Levels. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158674. [Google Scholar] [CrossRef]
- Reeves, H.L.; Friedman, S.L. Activation of Hepatic Stellate Cells—A Key Issue in Liver Fibrosis. Front. Biosci. 2002, 7, d808–d826. [Google Scholar] [CrossRef]
- Gandhi, C.R. Hepatic Stellate Cell Activation and Pro-Fibrogenic Signals. J. Hepatol. 2017, 67, 1104–1105. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Bataller, R.; Brenner, D.A. Human Hepatic Stellate Cells Express CCR5 and RANTES to Induce Proliferation and Migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G949–G958. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular Regulation of Hepatic Fibrosis, an Integrated Cellular Response to Tissue Injury*. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental Models of Liver Fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Xu, L. Human Hepatic Stellate Cell Lines, LX-1 and LX-2: New Tools for Analysis of Hepatic Fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Taimr, P.; Higuchi, H.; Kocova, E.; Rippe, R.A.; Friedman, S.; Gores, G.J. Activated Stellate Cells Express the TRAIL Receptor-2/Death Receptor-5 and Undergo TRAIL-Mediated Apoptosis. Hepatology 2003, 37, 87–95. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira da Silva, B.; Alberici, L.C.; Ramos, L.F.; Silva, C.M.; da Silveira, M.B.; Dechant, C.R.P.; Friedman, S.L.; Sakane, K.K.; Gonçalves, L.R.; Moraes, K.C.M. Altered Global MicroRNA Expression in Hepatic Stellate Cells LX-2 by Angiotensin-(1–7) and MiRNA-1914–5p Identification as Regulator of pro-Fibrogenic Elements and Lipid Metabolism. Int. J. Biochem. Cell Biol. 2018, 98, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Zeng, X.; Li, X.-M.; Wang, T.-L.; Wang, B.-E. Pirfenidone Inhibits Carbon Tetrachloride- and Albumin Complex-Induced Liver Fibrosis in Rodents by Preventing Activation of Hepatic Stellate Cells. Clin. Exp. Pharmacol. Physiol. 2009, 36, 963–968. [Google Scholar] [CrossRef]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. MBoC 2001, 12, 2730–2741. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nouchi, T.; Yamane, M.; Irie, T.; Miyakawa, H.; Sato, C.; Marumo, F. Phenotypic Modulation in Lipocytes in Experimental Liver Fibrosis. J. Pathol. 1991, 164, 273–278. [Google Scholar] [CrossRef]
- Hautekeete, M.L.; Geerts, A. The Hepatic Stellate (Ito) Cell: Its Role in Human Liver Disease. Virchows Archiv 1997, 430, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Suzuki, S.; Senoo, H. Hepatic Stellate Cells: Unique Characteristics in Cell Biology and Phenotype. Cell Struct. Funct. 2003, 28, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, B.; Hellerbrand, C.; Holcik, M.; Briendl, M.; Liebhaber, S.A.; Brenner, D.A. Posttranscriptional Regulation of Collagen alpha1(I) MRNA in Hepatic Stellate Cells. Mol. Cell. Biol. 1997, 17, 9. [Google Scholar] [CrossRef]
- Varela-Rey, M.; Montiel-Duarte, C.; Osés-Prieto, J.A.; López-Zabalza, M.J.; Jaffrèzou, J.P.; Rojkind, M.; Iraburu, M.J. P38 MAPK Mediates the Regulation of A1(I) Procollagen MRNA Levels by TNF-α and TGF-β in a Cell Line of Rat Hepatic Stellate Cells11The Opinions or Assertions Contained Herein Are the Private Views of the Authors and Are Not to Be Construed as Official or as Reflecting the Views of the Department of the Army or the Department of Defense of the US. FEBS Lett. 2002, 528, 133–138. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic Stellate Cell Lipid Droplets: A Specialized Lipid Droplet for Retinoid Storage. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of Retinyl Esters by Polyunsaturated Triacylglycerol Species in Lipid Droplets of Hepatic Stellate Cells during Activation. PLoS ONE 2012, 7, e34945. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Dunning, S.; ur Rehman, A.; Tiebosch, M.H.; Hannivoort, R.A.; Haijer, F.W.; Woudenberg, J.; van den Heuvel, F.A.J.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Glutathione and Antioxidant Enzymes Serve Complementary Roles in Protecting Activated Hepatic Stellate Cells against Hydrogen Peroxide-Induced Cell Death. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 2027–2034. [Google Scholar] [CrossRef]
- Byron, A.; Humphries, J.D.; Humphries, M.J. Defining the Extracellular Matrix Using Proteomics. Int. J. Exp. Pathol. 2013, 94, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Kovarova, M.; Feil, S.; Feil, R.; Wagner, R.; Heni, M.; Königsrainer, A.; Ruoß, M.; Nüssler, A.K.; Weigert, C.; et al. CGMP-Dependent Protein Kinase I (CGKI) Modulates Human Hepatic Stellate Cell Activation. Metabolism 2018, 88, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Liu, Y.; Zhang, Y.; Sun, Y.; Sun, C.; Zhang, Y.; Lv, X. Expression of Neuropeptide Y Is Increased in an Activated Human HSC Cell Line. Sci. Rep. 2019, 9, 9500. [Google Scholar] [CrossRef] [PubMed]
- Salhab, A.; Amer, J.; Lu, Y.; Safadi, R. Sodium+/Taurocholate Cotransporting Polypeptide as Target Therapy for Liver Fibrosis. Gut 2021. [Google Scholar] [CrossRef]
- Silva, C.M.; Ferrari, G.D.; Alberici, L.C.; Malaspina, O.; Moraes, K.C.M. Cellular and Molecular Effects of Silymarin on the Transdifferentiation Processes of LX-2 Cells and Its Connection with Lipid Metabolism. Mol. Cell Biochem. 2020, 468, 129–142. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, R.; Song, Z.; Ye, S.; Sun, R.; Xue, Q.; Zhang, Z. Gene Expression Profiles during Activation of Cultured Rat Hepatic Stellate Cells by Tumoral Hepatocytes and Fetal Bovine Serum. J. Cancer Res. Clin. Oncol. 2009, 136, 309. [Google Scholar] [CrossRef]
- Yuan, B.; Chen, Y.; Wu, Z.; Zhang, L.; Zhuang, Y.; Zhao, X.; Niu, H.; Cheng, J.C.-H.; Zeng, Z. Proteomic Profiling of Human Hepatic Stellate Cell Line LX2 Responses to Irradiation and TGF-β1. J. Proteome Res. 2019, 18, 508–521. [Google Scholar] [CrossRef]
- Ji, J.; Yu, F.; Ji, Q.; Li, Z.; Wang, K.; Zhang, J.; Lu, J.; Chen, L.; Qun, E.; Zeng, Y.; et al. Comparative Proteomic Analysis of Rat Hepatic Stellate Cell Activation: A Comprehensive View and Suppressed Immune Response. Hepatology 2012, 56, 332–349. [Google Scholar] [CrossRef]
- Ouyang, Y.; Guo, J.; Lin, C.; Lin, J.; Cao, Y.; Zhang, Y.; Wu, Y.; Chen, S.; Wang, J.; Chen, L.; et al. Transcriptomic Analysis of the Effects of Toll-like Receptor 4 and Its Ligands on the Gene Expression Network of Hepatic Stellate Cells. Fibrogenesis Tissue Repair 2016, 9, 2. [Google Scholar] [CrossRef]
- Xu, W.-H.; Hu, H.-G.; Tian, Y.; Wang, S.-Z.; Li, J.; Li, J.-Z.; Deng, X.; Qian, H.; Qiu, L.; Hu, Z.-L.; et al. Bioactive Compound Reveals a Novel Function for Ribosomal Protein S5 in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Hepatology 2014, 60, 648–660. [Google Scholar] [CrossRef]
- Olsen, A.L.; Sackey, B.K.; Marcinkiewicz, C.; Boettiger, D.; Wells, R.G. Fibronectin Extra Domain-A Promotes Hepatic Stellate Cell Motility but Not Differentiation into Myofibroblasts. Gastroenterology 2012, 142, 928.e3–937.e3. [Google Scholar] [CrossRef] [PubMed]
- Vogt, F.; Zernecke, A.; Beckner, M.; Krott, N.; Bosserhoff, A.-K.; Hoffmann, R.; Zandvoort, M.A.M.J.; Jahnke, T.; Kelm, M.; Weber, C.; et al. Blockade of Angio-Associated Migratory Cell Protein Inhibits Smooth Muscle Cell Migration and Neointima Formation in Accelerated Atherosclerosis. J. Am. Coll. Cardiol. 2008, 52, 302–311. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog Controls Hepatic Stellate Cell Fate by Regulating Metabolism. Gastroenterology 2012, 143, 1319.e11–1329.e11. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Basta, B.; Mato, J.M.; Craig, A.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Tsvirkun, D.; Hayardeny, L.; Chandar, V.; Schwartz, R.E.; et al. Aramchol Downregulates Stearoyl CoA-Desaturase 1 in Hepatic Stellate Cells to Attenuate Cellular Fibrogenesis. JHEP Rep. 2021, 3, 100237. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A.; Lazou, J.-M.; de Bleser, P.; Wisse, E. Tissue Distribution, Quantitation and Proliferation Kinetics of Fat-Storing Cells in Carbon Tetrachloride–Injured Rat Liver. Hepatology 1991, 13, 1193–1202. [Google Scholar] [CrossRef]
- Amakawa, M.; Endo, Y. The Motility of Hepatic Ito Cells Can Be Acquired by Their Myofibroblastic Transformation. Arch. Histol. Cytol. 2002, 65, 169–178. [Google Scholar] [CrossRef][Green Version]
- Cerbón-Ambriz, J.; Cerbón-Solórzano, J.; Rojkind, M. Regulation of Collagen Production in Freshly Isolated Cell Populations from Normal and Cirrhotic Rat Liver: Effect of Lactate. Hepatology 1991, 13, 551–556. [Google Scholar] [CrossRef]
- Kang, Q.; Chen, A. Curcumin Suppresses Expression of Low-density Lipoprotein (LDL) Receptor, Leading to the Inhibition of LDL-induced Activation of Hepatic Stellate Cells. Br. J. Pharmacol. 2009, 157, 1354–1367. [Google Scholar] [CrossRef]
- Bach Kristensen, D.; Kawada, N.; Imamura, K.; Miyamoto, Y.; Tateno, C.; Seki, S.; Kuroki, T.; Yoshizato, K. Proteome Analysis of Rat Hepatic Stellate Cells. Hepatology 2000, 32, 268–277. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, F.; Fan, X.; Lin, C.; Hao, Y.; Wei, H.; Lin, W.; Jiang, Y.; He, F. Quantitative Proteomic Analysis on Activated Hepatic Stellate Cells Reversion Reveal STAT1 as a Key Regulator between Liver Fibrosis and Recovery. Sci. Rep. 2017, 7, 44910. [Google Scholar] [CrossRef]
- Hamid, M.; Liu, D.; Abdulrahim, Y.; Liu, Y.; Qian, G.; Khan, A.; Gan, F.; Huang, K. Amelioration of CCl4-Induced Liver Injury in Rats by Selenizing Astragalus Polysaccharides: Role of Proinflammatory Cytokines, Oxidative Stress and Hepatic Stellate Cells. Res. Vet. Sci. 2017, 114, 202–211. [Google Scholar] [CrossRef]
- Kim, M.-B.; Bae, M.; Hu, S.; Kang, H.; Park, Y.-K.; Lee, J.-Y. Fucoxanthin Exerts Anti-Fibrogenic Effects in Hepatic Stellate Cells. Biochem. Biophys. Res. Commun. 2019, 513, 657–662. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A Practical Guide to the MaxQuant Computational Platform for SILAC-Based Quantitative Proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Model in Literature | Results in Literature | Results in This Study | References |
|---|---|---|---|---|
| Proliferation | Primary rat HSC | Increase of HSC numbers after CCl4 injury (activation) | Increased proliferation after serum activation, upregulation of ribosome biogenesis and cell cycle proteins | [45] |
| Migration | Primary rat HSC | Increased motility after PDGF-β activation | Increased migration after serum activation, upregulation of migration associated proteins | [46] |
| ECM production (collagen) | Primary rat HSC | Strong upregulation of collagen production in HSC CCl4 treatment (activation) | Increased COL1A1 production in serum activated LX-2, upregulation of ECM regulators and ER-Golgi transport | [47] |
| Lipid metabolism | Primary rat HSC | LDs increase in number, but decrease in total volume during activation | LDs decrease in volume, downregulation of proteins involved in lipid biosynthesis after serum activation | [27] |
| Cholesterol metabolism | Primary rat HSC, primary human HSC & LX-2 | Downregulation of cholesterol biosynthesis in HSC ameliorates liver fibrosis | Downregulation of proteins involved in cholesterol biosynthesis in serum activated LX-2 | [44,48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schinagl, M.; Tomin, T.; Gindlhuber, J.; Honeder, S.; Pfleger, R.; Schittmayer, M.; Trauner, M.; Birner-Gruenberger, R. Proteomic Changes of Activated Hepatic Stellate Cells. Int. J. Mol. Sci. 2021, 22, 12782. https://doi.org/10.3390/ijms222312782
Schinagl M, Tomin T, Gindlhuber J, Honeder S, Pfleger R, Schittmayer M, Trauner M, Birner-Gruenberger R. Proteomic Changes of Activated Hepatic Stellate Cells. International Journal of Molecular Sciences. 2021; 22(23):12782. https://doi.org/10.3390/ijms222312782
Chicago/Turabian StyleSchinagl, Maximilian, Tamara Tomin, Juergen Gindlhuber, Sophie Honeder, Raphael Pfleger, Matthias Schittmayer, Michael Trauner, and Ruth Birner-Gruenberger. 2021. "Proteomic Changes of Activated Hepatic Stellate Cells" International Journal of Molecular Sciences 22, no. 23: 12782. https://doi.org/10.3390/ijms222312782
APA StyleSchinagl, M., Tomin, T., Gindlhuber, J., Honeder, S., Pfleger, R., Schittmayer, M., Trauner, M., & Birner-Gruenberger, R. (2021). Proteomic Changes of Activated Hepatic Stellate Cells. International Journal of Molecular Sciences, 22(23), 12782. https://doi.org/10.3390/ijms222312782