
PTP61F Mediates Cell Competition and Mitigates Tumorigenesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
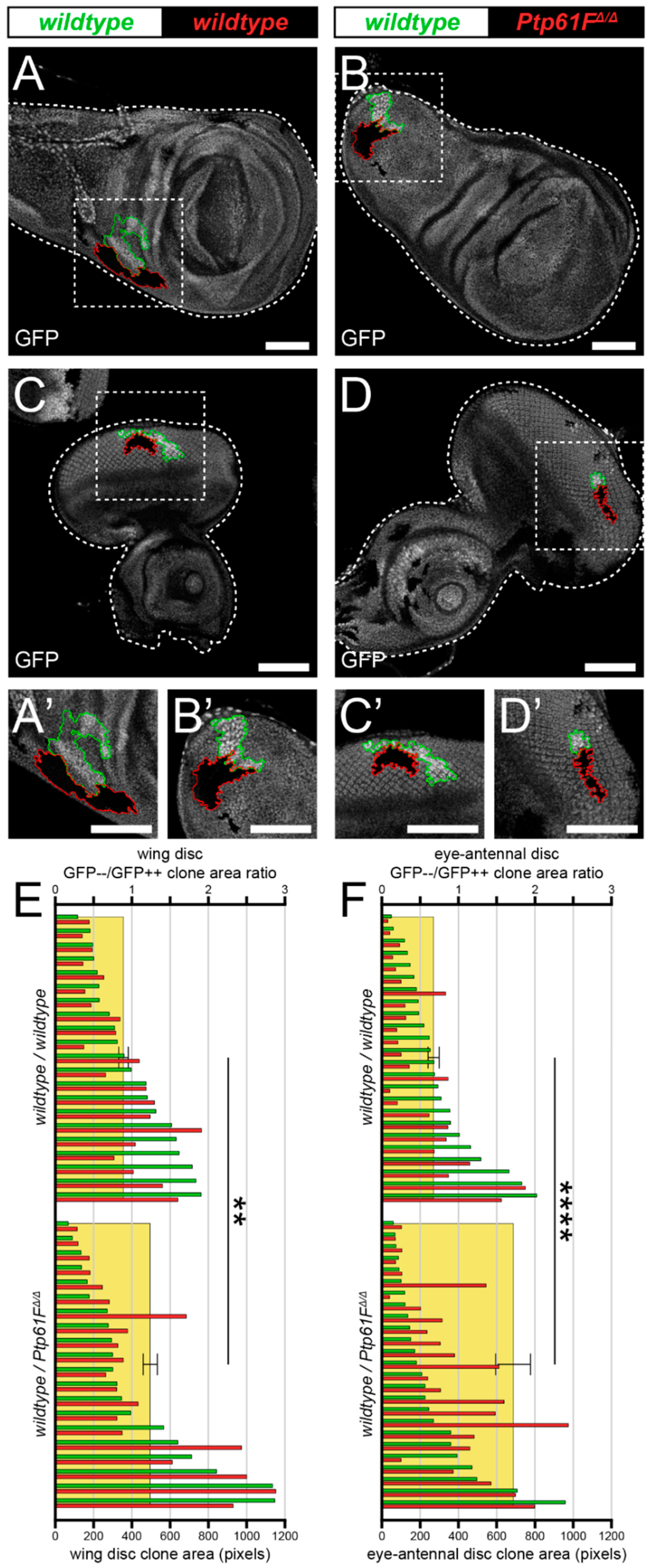
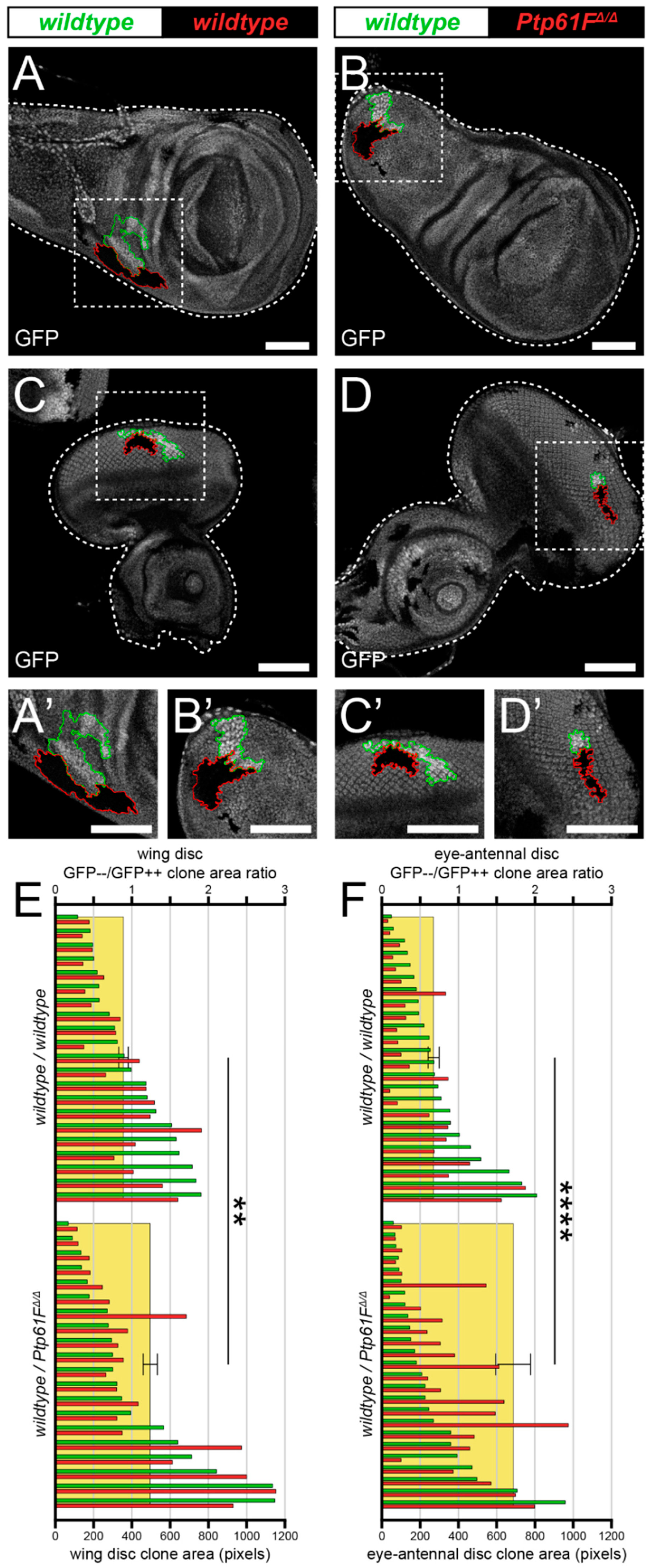
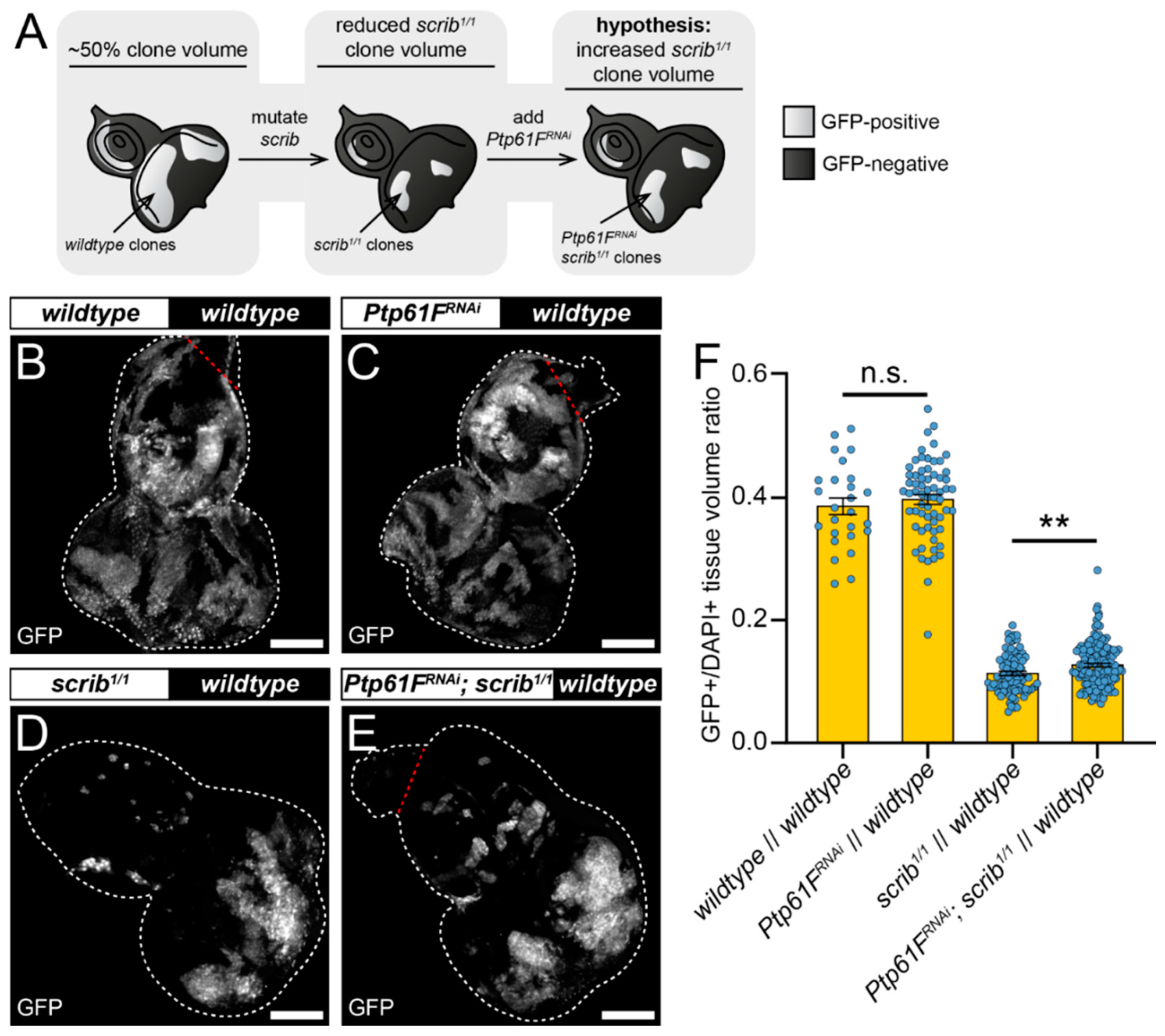
2.1. Ptp61F Impairment Confers a Competitive Advantage on Epithelial Clones
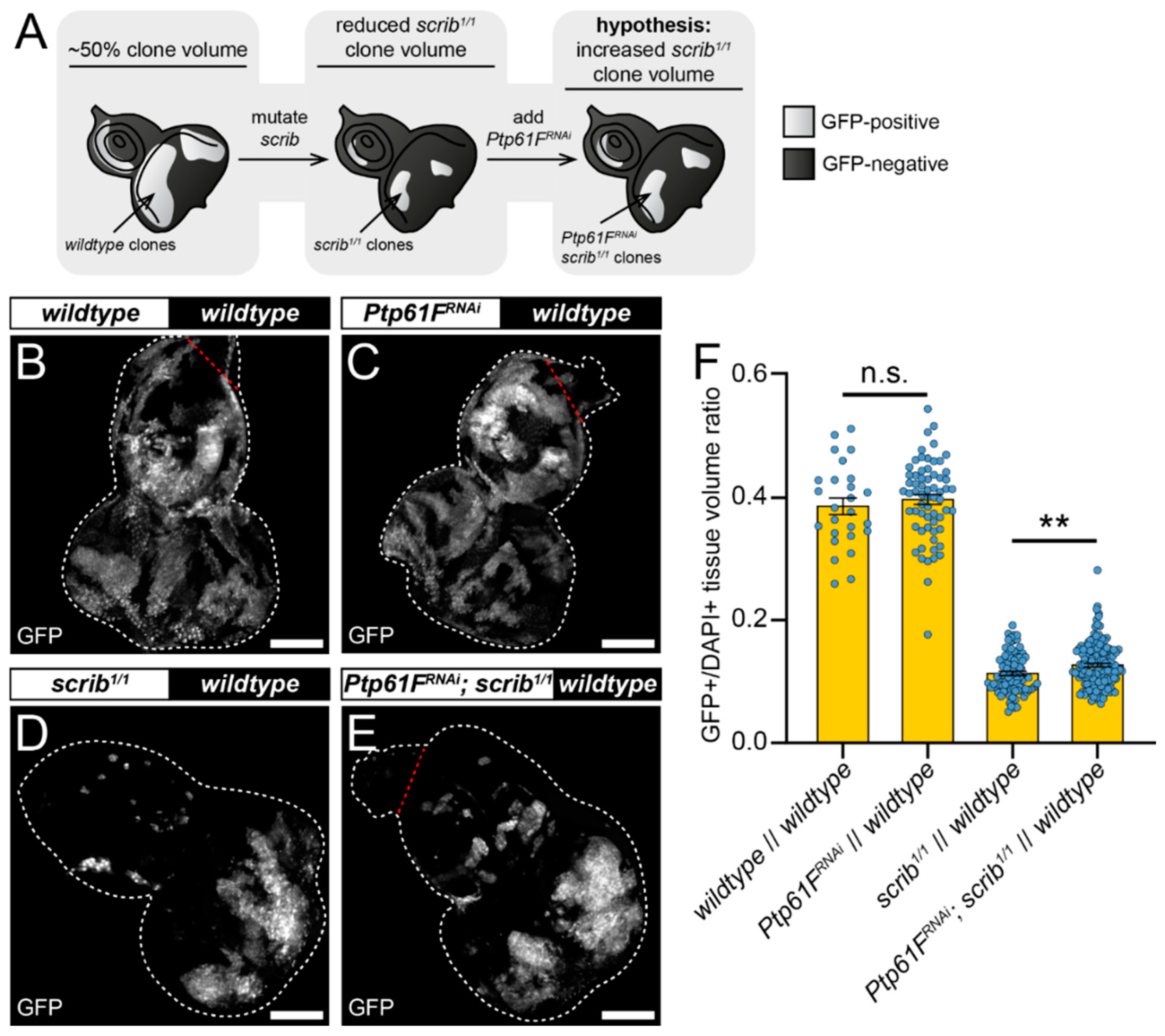
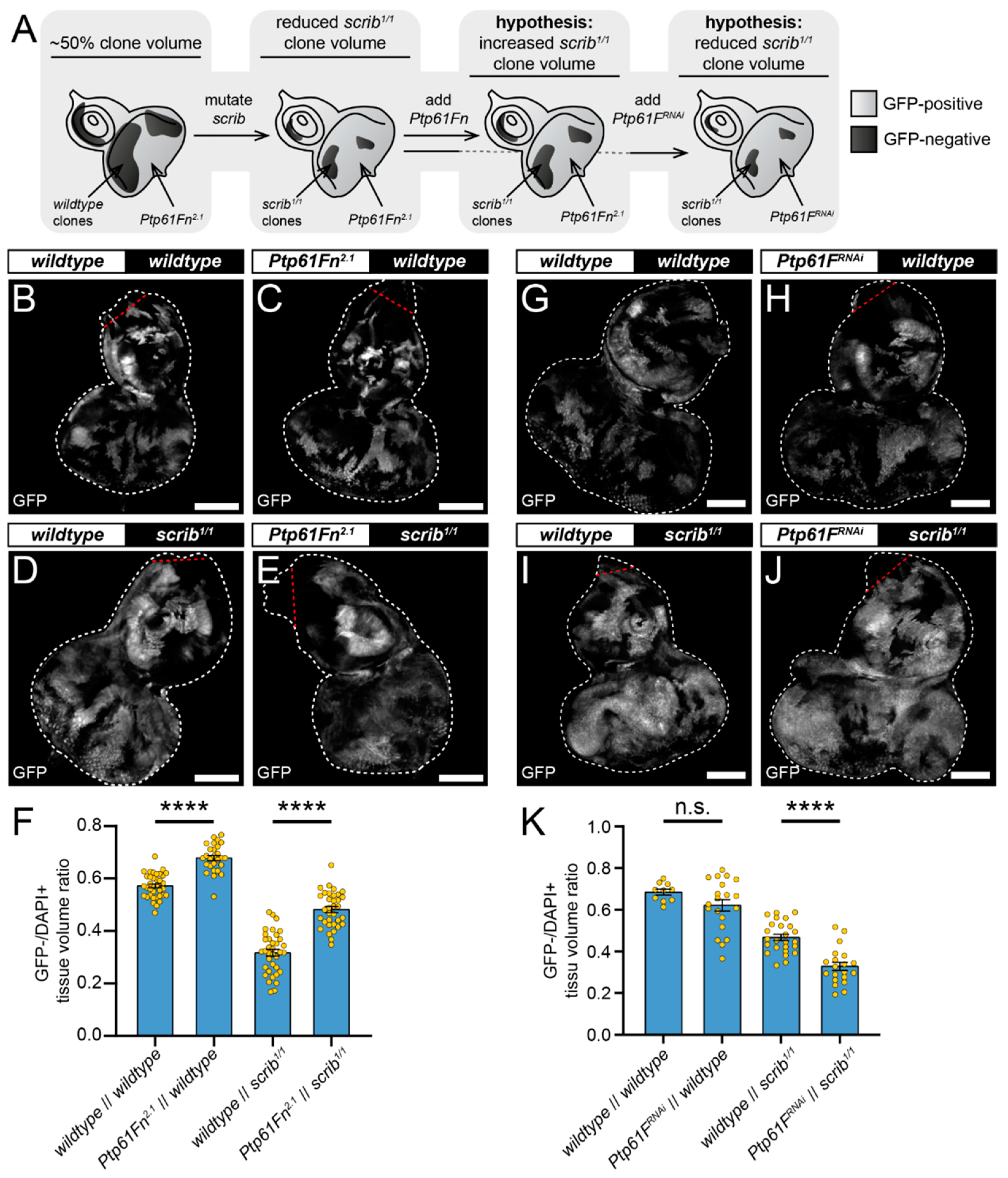
2.2. Ptp61F Regulates Polarity-Impaired Clone Survival/Growth during Cell Competition
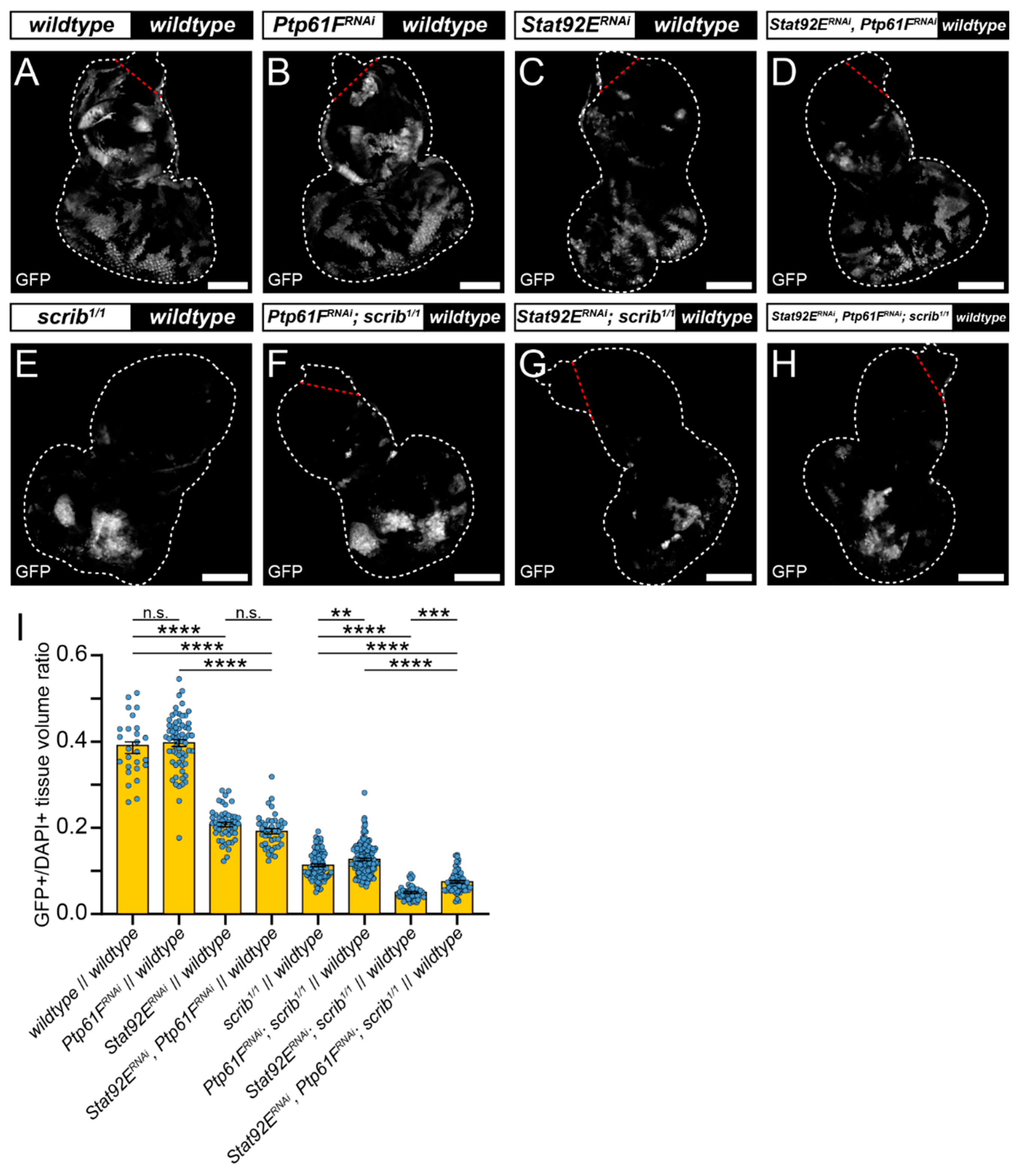
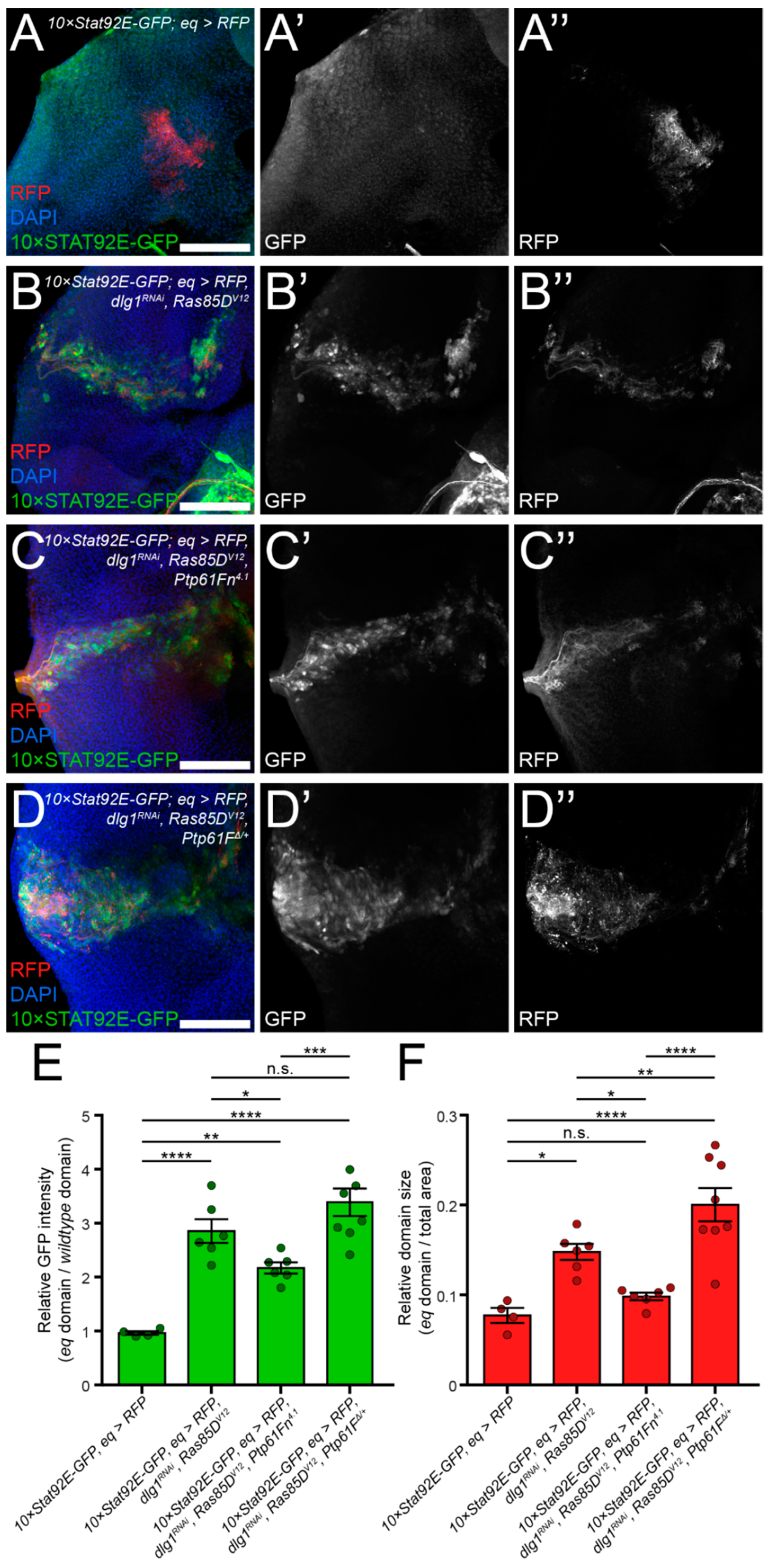
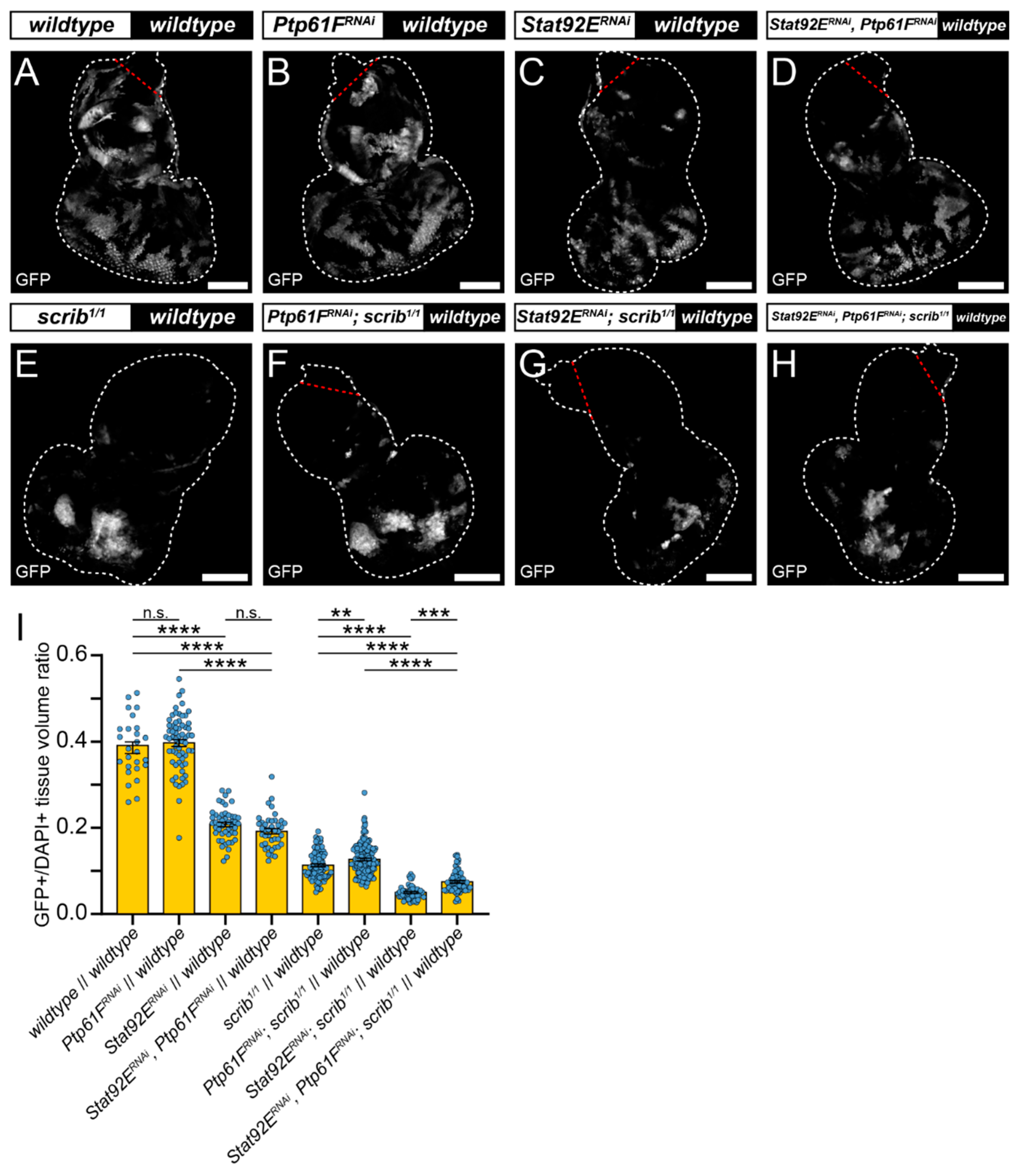
2.3. JAK–STAT Signalling Plays a Role in the Fitness of Scrib-Mutant Clones and Is Required Downstream of Ptp61F Knockdown for the Increased Survival of Scrib-Mutant Clones
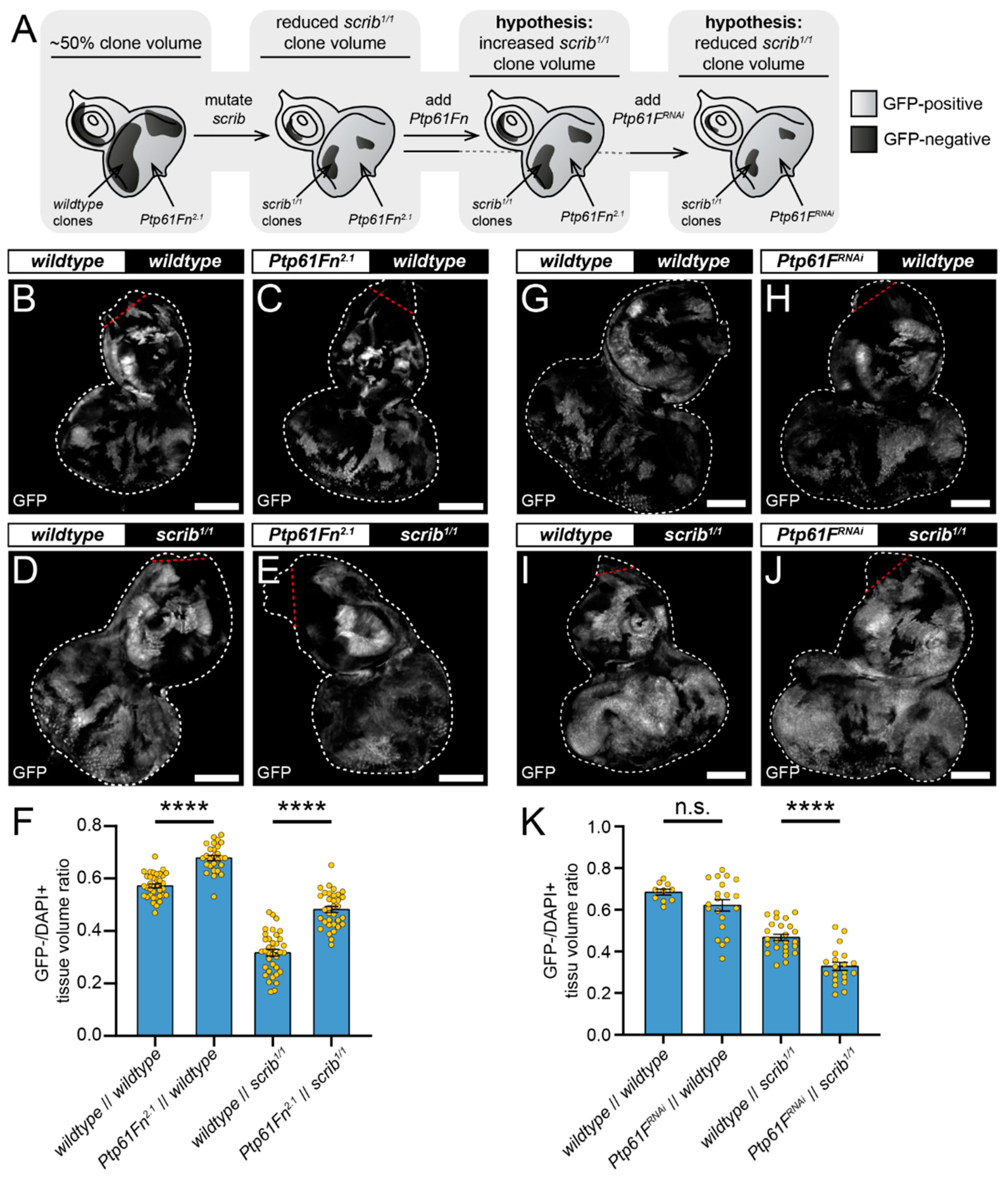
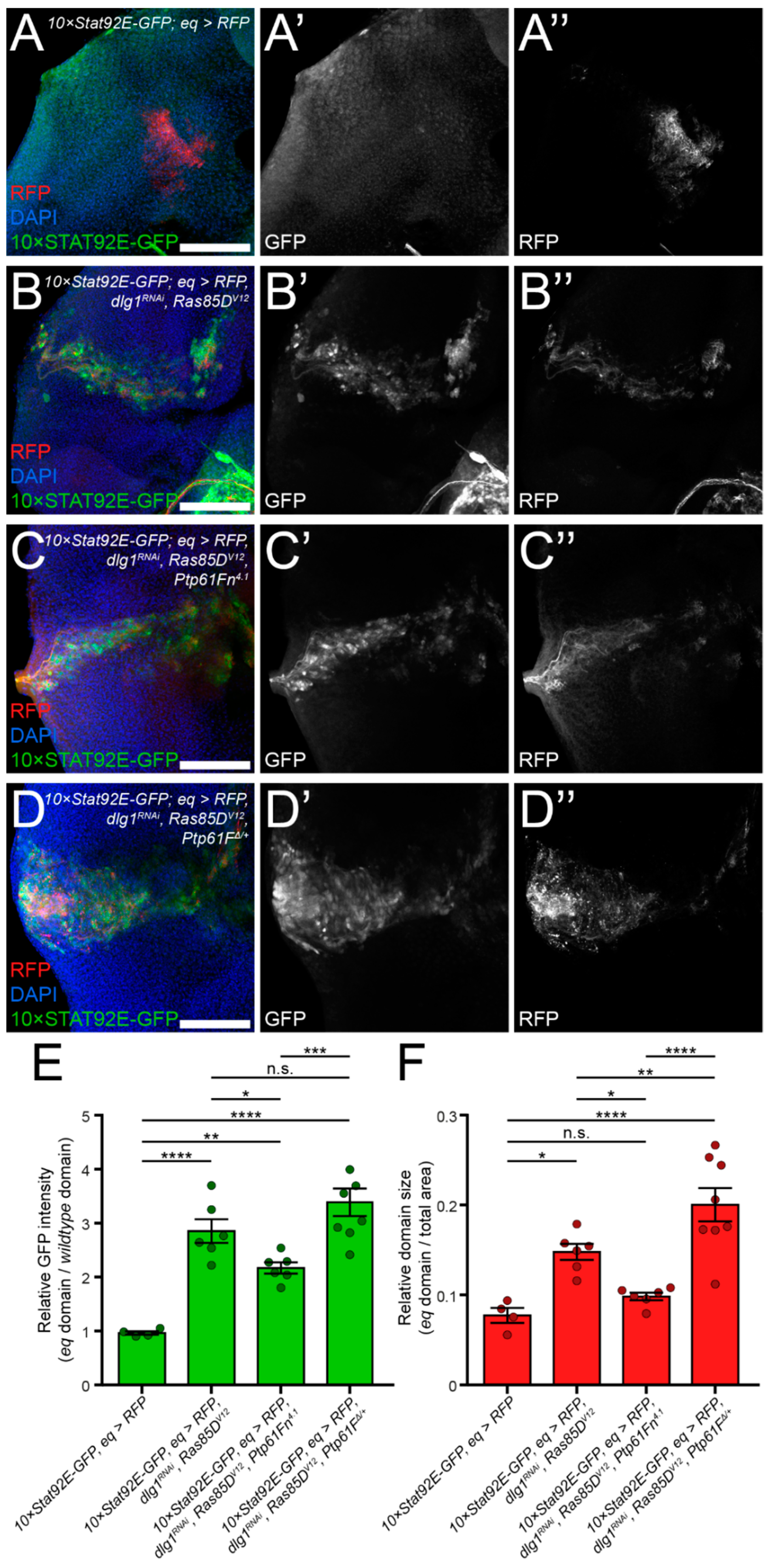
2.4. Ptp61F Regulates Wild-Type Clone-Mediated Elimination of Polarity-Impaired Clones during Cell Competition
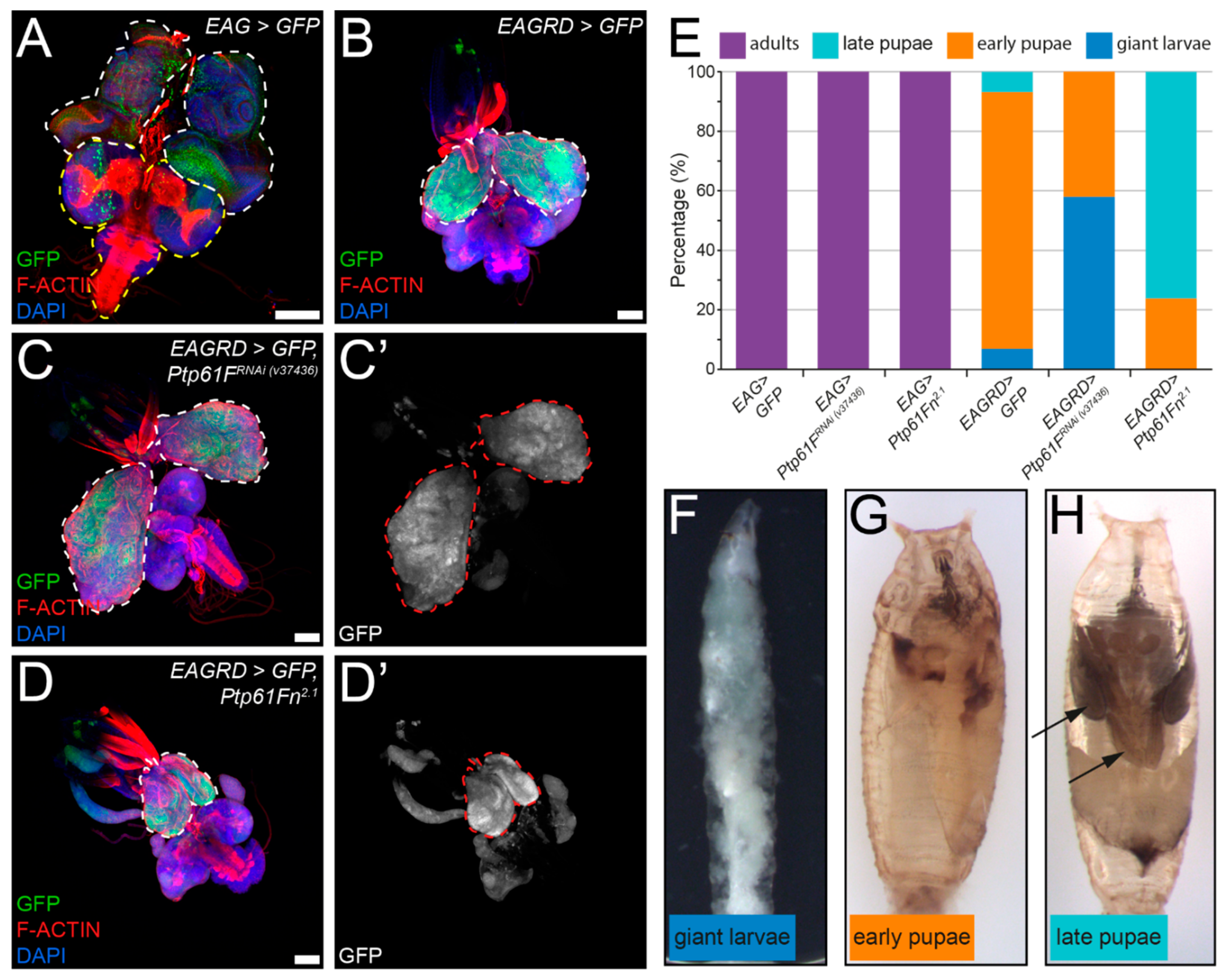
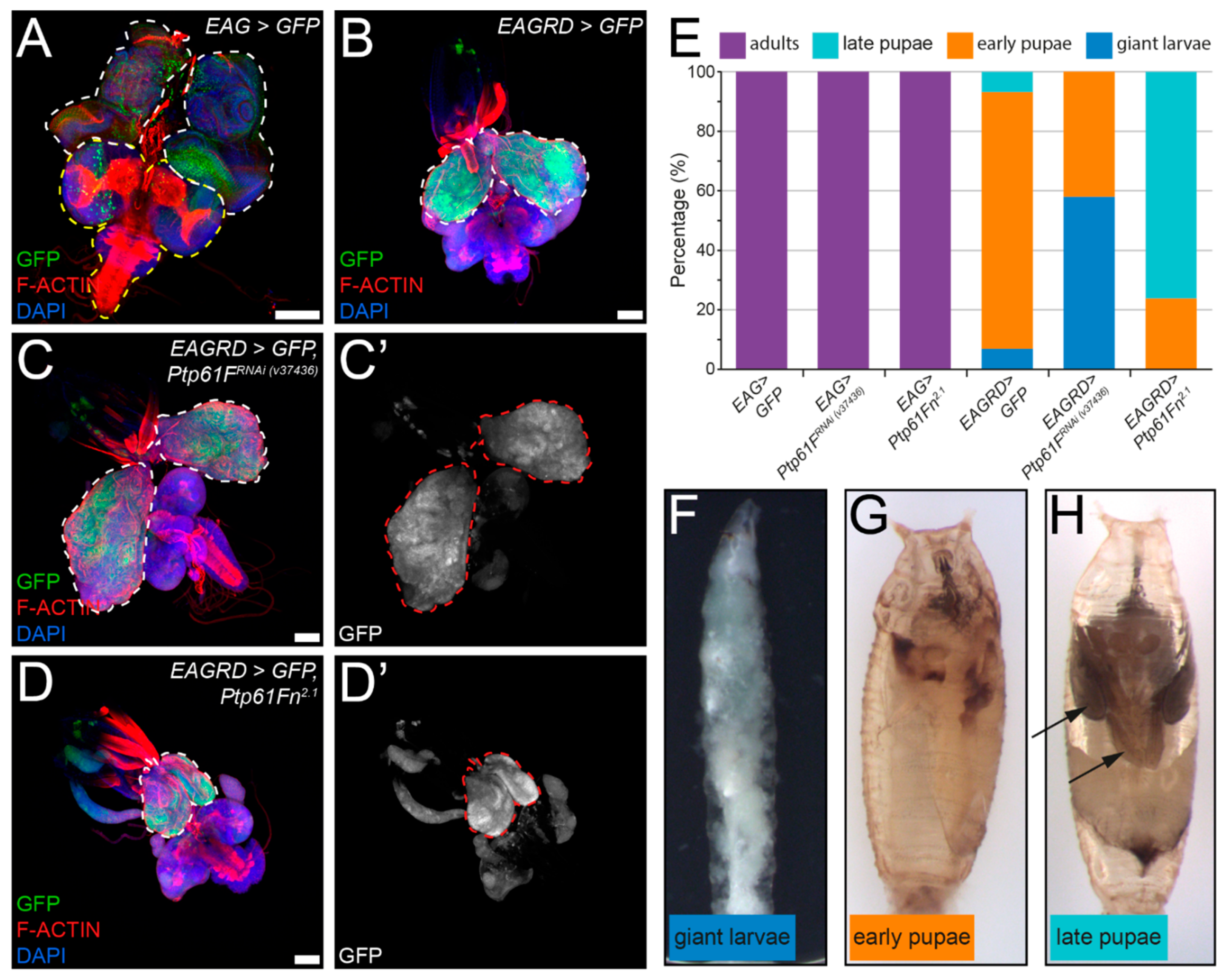
2.5. Ptp61F Suppresses Activated Ras85D-Driven Polarity-Impaired Epithelial Tumorigenesis
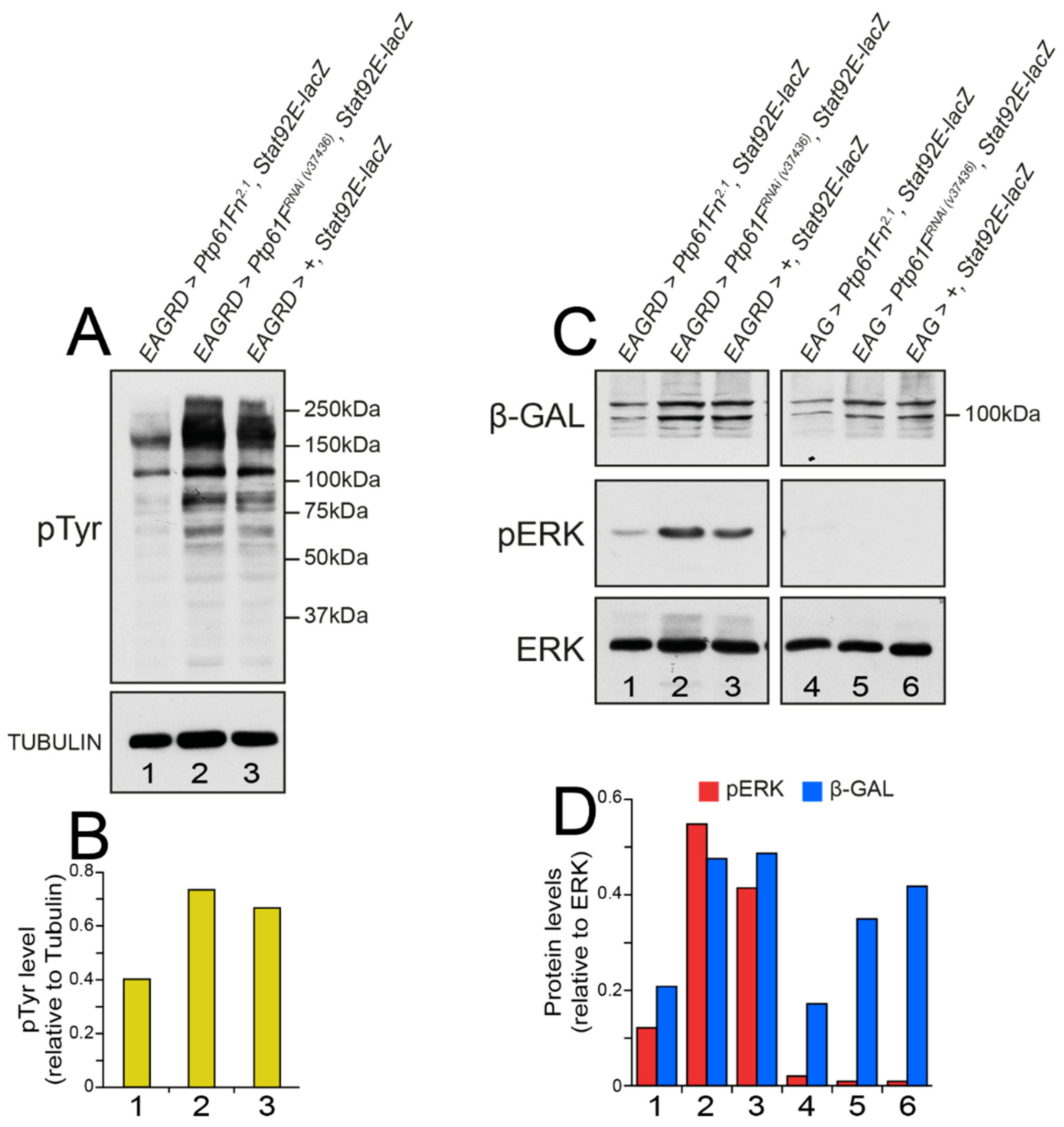
2.6. Drosophila Ptp61F Represses pTyr, RAS–MAPK, and JAK–STAT Signalling in Activated Ras85D-Driven Polarity-Impaired Tumors
3. Discussion
3.1. Summary of the Result of This Study
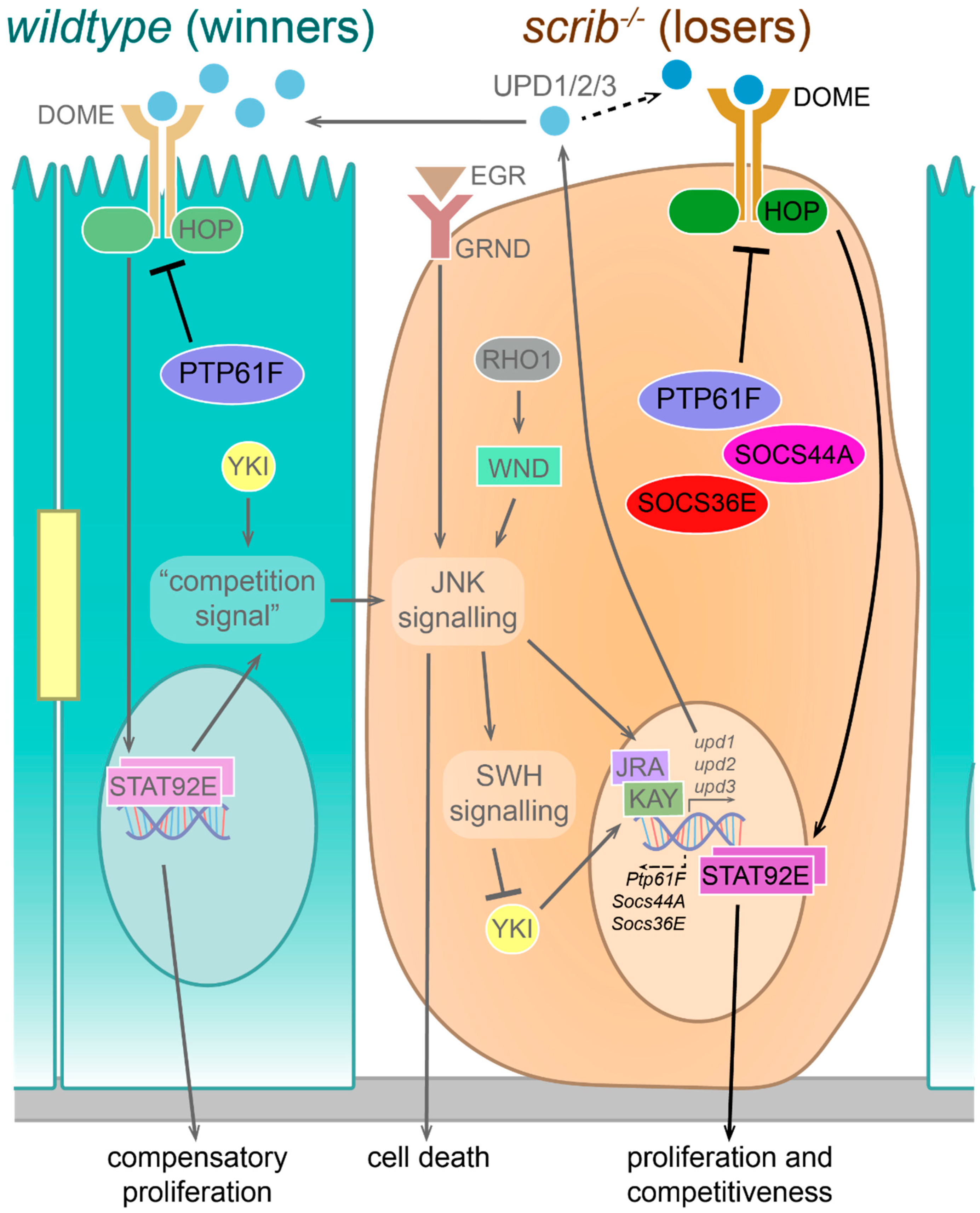
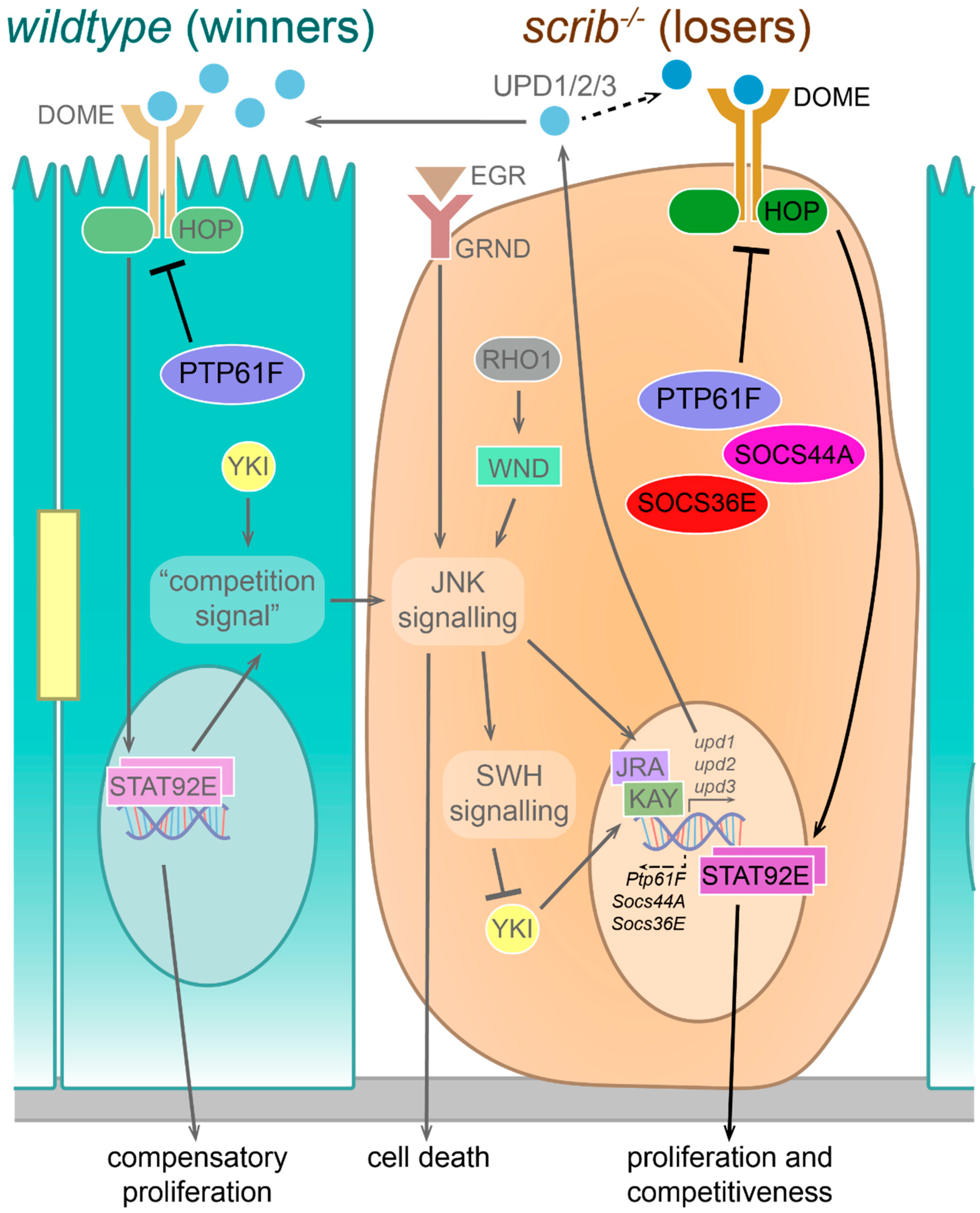
3.2. PTP61F in Signalling Pathway Regulation during Cell Competition
3.3. Human Orthologs of PTP61F in Tumorigenesis
4. Materials and Methods
4.1. Fly Stocks and General Husbandry
4.2. Imaging and Sample Preparation
4.3. Twin-Clone Generation and Quantification
4.4. MARCM and Reverse MARCM
4.5. Clone Volume Quantification and Statistical Analyses
4.6. Statistical Analyses of Signal Intensity and the eq Expression Domain Area
4.7. Western Blotting, Analyses, and Antibodies
4.8. RNA Extraction, cDNA Synthesis, and qRT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, L.Y.; Parsons, L.M.; Richardson, H.E. Modelling Cancer in Drosophila: The Next Generation. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2013. [Google Scholar]
- Gonzalez, C. Drosophila melanogaster: A model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 2013, 13, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Sonoshita, M.; Cagan, R.L. Modeling Human Cancers in Drosophila. In Current Topics in Developmental Biology—Fly Models of Human Diseases; Pick, L., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 287–309. [Google Scholar]
- Richardson, H.E.; Portela, M. Modelling Cooperative Tumorigenesis in Drosophila. BioMed Res. Int. 2018, 2018, 4258387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Marca, J.E.; Richardson, H.E. Two-Faced: Roles of JNK Signalling During Tumourigenesis in the Drosophila Model. Front. Cell Dev. Biol. 2020, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, F.D.; Rubin, G.M. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 1998, 125, 1–9. [Google Scholar] [CrossRef]
- Pagliarini, R.A.; Xu, T. A Genetic Screen in Drosophila for Metastatic Behavior. Science 2003, 302, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.; Li, M.; Perrimon, N. Cooperative Regulation of Cell Polarity and Growth by Drosophila Tumor Suppressors. Science 2000, 289, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Bilder, D.; Perrimon, N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature 2000, 403, 676–680. [Google Scholar] [CrossRef]
- Brumby, A.M.; Richardson, H.E. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef]
- Leong, G.R.; Goulding, K.R.; Amin, N.; Richardson, H.E.; Brumby, A.M. Scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol. 2009, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Xu, W.; Zhang, D.; Yang, Y.; Li, W.; Xue, L. Wallenda regulates JNK-mediated cell death in Drosophila. Cell Death Dis. 2015, 6, 1737. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of Cell Polarity Drives Tumor Growth and Invasion through JNK Activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Uhlirova, M.; Jasper, H.; Bohmann, D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc. Natl. Acad. Sci. USA 2005, 102, 13123–13128. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Schroeder, M.; Kango-Singh, M.; Tao, C.; Halder, G. Tumor suppression by cell competition through regulation of the Hippo pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey-Lozano, N.; La Marca, J.E.; Portela, M.; Richardson, H.E. Drosophila Models of Cell Polarity and Cell Competition in Tumourigenesis. In Advances in Experimental Medicine and Biology—The Drosophila Model in Cancer; Deng, W.-M., Ed.; Springer: Cham, Switzerland, 2019; pp. 36–74. [Google Scholar]
- Madan, E.; Gogna, R.; Moreno, E. Cell competition in development: Information from flies and vertebrates. Curr. Opin. Cell Biol. 2018, 55, 150–157. [Google Scholar] [CrossRef]
- Cohen, P.T.W. Novel protein serine/threonine phosphatases: Variety is the spice of life. Trends Biochem. Sci. 1997, 22, 245–251. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [Green Version]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Hatzihristidis, T.; Liu, S.; Pyszcz, L.; Hutchins, A.P.; Gabaldon, T.; Tremblay, M.; Miranda-Saavedra, D. PTP-central: A comprehensive resource of protein tyrosine phosphatases in eukaryotic genomes. Methods 2014, 65, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K.; Murakami, M.S.; Cleghon, V. Protein Kinases and Phosphatases in the Drosophila Genome. J. Cell Biol. 2000, 150, 57–62. [Google Scholar] [CrossRef]
- Thurmond, J.; Goodman, J.L.; Strelets, V.B.; Attrill, H.; Gramates, L.S.; Marygold, S.J.; Matthews, B.B.; Millburn, G.; Antonazzo, G.; Trovisco, V.; et al. FlyBase 2.0: The next generation. Nucleic Acids Res. 2018, 47, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Buszard, B.J.; Johnson, T.K.; Meng, T.-C.; Burke, R.; Warr, C.G.; Tiganis, T. The Nucleus- and Endoplasmic Reticulum-Targeted Forms of Protein Tyrosine Phosphatase 61F Regulate Drosophila Growth, Life Span, and Fecundity. Mol. Cell. Biol. 2013, 33, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, P.; Kuttenkeuler, D.; Gesellchen, V.; Zeidler, M.P.; Boutros, M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 2005, 436, 871–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadin, A.; Starz-Gaiano, M. Identification of Novel Regulators of the JAK/STAT Signaling Pathway that Control Border Cell Migration in the Drosophila Ovary. G3 Genes Genomes Genet. 2016, 6, 1991–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, L.F.; Manent, J.; Allan, K.; Lee, H.; Portela, M.; Wiede, F.; Warr, C.; Meng, T.-Z.; Tiganis, T.; Richardson, H.E. Differential regulation of protein tyrosine kinase signalling by Dock and the PTP61F variants. FEBS J. 2017, 284, 2231–2250. [Google Scholar] [CrossRef] [Green Version]
- Tchankouo-Nguetcheu, S.; Udinotti, M.; Durand, M.; Meng, T.Z.; Taouis, M.; Rabinow, L. Negative regulation of MAP kinase signaling in Drosophila by Ptp61F/PTP1B. Mol. Genet. Genom. 2014, 289, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-L.; Buszard, B.; Teng, C.H.; Chen, W.L.; Warr, C.G.; Tiganis, T.; Meng, T.C. Dock/Nck facilitates PTP61F/PTP1B regulation of insulin signalling. Biochem. J. 2011, 439, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Hatzihristidis, T.; Desai, N.; Hutchins, A.P.; Meng, T.-C.; Tremblay, M.; Miranda-Saavedra, D. A Drosophila-centric view of protein tyrosine phosphatases. FEBS Lett. 2015, 589, 951–966. [Google Scholar] [CrossRef] [Green Version]
- Tiganis, T. PTP1B and TCPTP—Nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013, 280, 445–458. [Google Scholar] [CrossRef]
- Tonks, N.K.; Diltz, C.D.; Fischer, E.H. Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6722–6730. [Google Scholar] [CrossRef]
- Elchebly, M. Increased Insulin Sensitivity and Obesity Resistance in Mice Lacking the Protein Tyrosine Phosphatase-1B Gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Galic, S.; Hauser, C.; Kahn, B.B.; Haj, F.G.; Neel, B.G.; Tonks, N.K.; Tiganis, T. Coordinated Regulation of Insulin Signaling by the Protein Tyrosine Phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 2005, 25, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Klaman, L.D.; Boss, O.; Perroni, O.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.; et al. Increased Energy Expenditure, Decreased Adiposity, and Tissue-Specific Insulin Sensitivity in Protein-Tyrosine Phosphatase 1B-Deficient Mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.P. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 2001, 276, 47771–47774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Bjorge, J.D.; Fujita, D.J. PTP1B Contributes to the Oncogenic Properties of Colon Cancer Cells through Src Activation. Cancer Res. 2007, 67, 10129–10137. [Google Scholar] [CrossRef] [Green Version]
- Arias-Romero, L.E.; Saha, S.; Villamar-Cruz, O.; Yip, S.C.; Ethier, S.P.; Zhang, Z.-Y.; Chernoff, J. Activation of Src by Protein Tyrosine Phosphatase 1B Is Required for ErbB2 Transformation of Human Breast Epithelial Cells. Cancer Res. 2009, 69, 4582–4588. [Google Scholar] [CrossRef] [Green Version]
- Bentires-Alj, M.; Neel, B.G. Protein-Tyrosine Phosphatase 1B Is Required for HER2/Neu–Induced Breast Cancer. Cancer Res. 2007, 67, 2420–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, S.G.; Dube, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.; Tremblay, M. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.A.; Dadabay, C.Y.; Fischer, E.H. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J. Cell Biol. 1995, 131, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Mosinger, B.; Tillmann, U.; Westphal, H.; Tremblay, M.L. Cloning and characterization of a mouse cDNA encoding a cytoplasmic protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 1992, 89, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Tiganis, T.; Flint, A.J.; Adam, S.A.; Tonks, N.K. Association of the T-cell Protein Tyrosine Phosphatase with Nuclear Import Factor p97. J. Biol. Chem. 1997, 272, 21548–21557. [Google Scholar] [CrossRef] [Green Version]
- Tiganis, T.; Bennett, A.M. Protein tyrosine phosphatase function: The substrate perspective. Biochem. J. 2007, 402, 1–15. [Google Scholar] [CrossRef]
- Kleppe, M.; Lahortiga, I.; El Chaar, T.; De Keersmaecker, K.; Mentens, N.; Graux, C.; Van Roosbroeck, K.; Ferrardo, A.A.; Langerak, A.; Meijerink, J.P.P.; et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 530–535. [Google Scholar] [CrossRef]
- Kleppe, M.; Soulier, J.; Asnafi, V.; Mentens, N.; Hornakova, T.; Knoops, L.; Constantinescu, S.; Sigaux, F.; Meijerink, J.P.; Vandenberghe, P.; et al. PTPN2 negatively regulates oncogenic JAK1 in T-cell acute lymphoblastic leukemia. Blood 2011, 117, 7090–7098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, B.J.; Wiede, F.; Gurzov, E.N.; Wee, K.; Hauser, C.; Zhu, H.-J.; Molloy, T.J.; O’Toole, S.A.; Daly, R.; Sutherland, R.L.; et al. TCPTP Regulates SFK and STAT3 Signaling and Is Lost in Triple-Negative Breast Cancers. Mol. Cell. Biol. 2013, 33, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiganis, T. The Role of TCPTP in Cancer. In Protein Tyrosine Phosphatases in Cancer; Neel, B.G., Tonks, N.K., Eds.; Springer: New York, NY, USA, 2016; pp. 145–168. [Google Scholar]
- Grohmann, M.; Wiede, F.; Dodd, G.; Gurzov, E.N.; Ooi, G.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froldi, F.; Ziosi, M.; Garoia, F.; Pession, A.; Grzeschik, N.A.; Bellosta, P.; Strand, D.; Richardson, H.E.; Pession, A.; Griffoni, D. The lethal giant larvae tumour suppressor mutation requires dMyc oncoprotein to promote clonal malignancy. BMC Biol. 2010, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Luo, L. Mosaic Analysis with a Repressible Cell Marker for Studies of Gene Function in Neuronal Morphogenesis. Neuron 1999, 22, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Baeg, G.-H.; Zhou, R.; Perrimon, N. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 2005, 19, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, M.C.; Chen, C.-L.; Gajewski, K.; Halder, G. A non-cell-autonomous tumor suppressor role for Stat in eliminating oncogenic scribble cells. Oncogene 2013, 32, 4471–4479. [Google Scholar] [CrossRef] [Green Version]
- Willecke, M.; Toggweiler, J.; Basler, K. Loss of PI3K blocks cell-cycle progression in a Drosophila tumor model. Oncogene 2011, 30, 4067–4074. [Google Scholar] [CrossRef] [Green Version]
- Casci, T.; Freeman, M. Control of EGF Receptor Signalling: Lessons from Fruitflies. Cancer Metastasis Rev. 1999, 18, 181–201. [Google Scholar] [CrossRef]
- Malartre, M. Regulatory mechanisms of EGFR signalling during Drosophila eye development. Cell. Mol. Life Sci. 2016, 73, 1825–1843. [Google Scholar] [CrossRef] [PubMed]
- Bunker, B.D. The transcriptional response to tumorigenic polarity loss in Drosophila. eLife 2015, 4, 03189. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between RasV12 and scribbled clones induces tumour growth and invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.; Yao, J.G.; Chen, P.-H.; Posakony, J.W.; Barolo, S.; Kim, J.; Sun, H. Upd/Jak/STAT signaling represses wg transcription to allow initiation of morphogenetic furrow in Drosophila eye development. Dev. Biol. 2007, 306, 760–771. [Google Scholar] [CrossRef] [Green Version]
- Bach, E.A.; Ekas, L.A.; Ayala-Camargo, A.; Flaherty, M.S.; Lee, H.; Perrimon, N.; Baeg, G.-H. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr. Patterns 2007, 7, 323–331. [Google Scholar] [CrossRef]
- Rodrigues, A.B.; Zoranovic, T.; Ayala-Camargo, A.; Grewal, S.; Reyes-Robles, T.; Krasny, M.; Wu, C.; Johnston, L.A.; Bach, E.A. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development 2012, 139, 4051–4061. [Google Scholar] [CrossRef] [Green Version]
- Andersen, D.S.; Colombani, J.; Palmerini, V.; Chakrabandhu, K.; Boone, E.; Röthlisberger, M.; Toggweiler, J.; Basler, K.; Mapelli, M.; Hueber, A.-O.; et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature 2015, 522, 482–486. [Google Scholar] [CrossRef]
- Ma, X.; Chen, Y.; Zhang, S.; Xu, W.; Shao, Y.; Li, W.; Li, M.; Xue, L. Rho1–Wnd signaling regulates loss-of-cell polarity-induced cell invasion in Drosophila. Oncogene 2016, 35, 846–855. [Google Scholar] [CrossRef]
- Igaki, T.; Pastor-Pareja, J.C.; Aonuma, H.; Miura, M.; Xu, T. Intrinsic Tumor Suppression and Epithelial Maintenance by Endocytic Activation of Eiger/TNF Signaling in Drosophila. Dev. Cell 2009, 16, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Herranz, H.; Hong, X.; Hung, N.T.; Voorhoeve, P.M.; Cohen, S. Oncogenic cooperation between SOCS family proteins and EGFR identified using a Drosophila epithelial transformation model. Genes Dev. 2012, 26, 1602–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar]
- Stec, W.J.; Zeidler, M.P. Drosophila SOCS Proteins. J. Signal Transduct. 2011, 2011, 894510. [Google Scholar] [CrossRef] [Green Version]
- Issigonis, M.; Matunis, E. The Drosophila BCL6 homolog ken and barbie promotes somatic stem cell self-renewal in the testis niche. Dev. Biol. 2012, 368, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S. Two Drosophila suppressors of cytokine signaling (SOCS) differentially regulate JAK and EGFR pathway activities. BMC Cell Biol. 2004, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Callus, B.A.; Mathey-Prevot, B. SOCS36E, a novel Drosophila SOCS protein, suppresses JAK/STAT and EGF-R signalling in the imaginal wing disc. Oncogene 2002, 21, 4812–4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsten, P.; Häder, S.; Zeidler, M.P. Cloning and expression of Drosophila SOCS36E and its potential regulation by the JAK/STAT pathway. Mech. Dev. 2002, 117, 343–346. [Google Scholar] [CrossRef]
- Labbé, D.P.; Tremblay, M.L. PTP1B: From Metabolism to Cancer. In Protein Tyrosine Phosphatases in Cancer; Neel, B.G., Tonks, N.K., Eds.; Springer: New York, NY, USA, 2016; pp. 169–200. [Google Scholar]
- Yamamoto, M.; Ohsawa, S.; Kunimasa, K.; Igaki, T. The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition. Nature 2017, 542, 246–250. [Google Scholar] [CrossRef]
- Portela, M.; Casas-Tinto, S.; Rhiner, C.; Lopez-Gay, J.M.; Dominguez, O.; Soldini, D.; Moreno, E. Drosophila SPARC Is a Self-Protective Signal Expressed by Loser Cells during Cell Competition. Dev. Cell 2010, 19, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Rhiner, C.; Lopez-Gay, J.M.; Soldini, D.; Casas-Tinto, C.; Martin, F.A.; Lombardia, L.; Moreno, E. Flower Forms an Extracellular Code that Reveals the Fitness of a Cell to its Neighbors in Drosophila. Dev. Cell 2010, 18, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Katsukawa, M.; Ohsawa , S.; Zhang , L.; Yan , Y.; Igaki , T. Serpin Facilitates Tumor-Suppressive Cell Competition by Blocking Toll-Mediated Yki Activation in Drosophila. Curr. Biol. 2018, 28, 1756–1767. [Google Scholar] [CrossRef] [Green Version]
- Le Sommer, S.; Ohsawa, S.; Zhang, L.; Yan, Y.; Igaki, T. Deficiency in Protein Tyrosine Phosphatase PTP1B Shortens Lifespan and Leads to Development of Acute Leukemia. Cancer Res. 2018, 78, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Lessard, L.; Labbe, D.P.; Deblois, G.; Begin, L.R.; Hardy, S.; Mes-Masson, A.M.; Saad, F.; Trotman, L.C.; Viguere, V.; Tremblay, M.L. PTP1B Is an Androgen Receptor–Regulated Phosphatase That Promotes the Progression of Prostate Cancer. Cancer Res. 2012, 72, 1529–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhang, L.; Bourne, P.A.; Reeder, J.E.; di Sant’Agnese, P.A.; Yao, J.; Na, Y.; Huang, J. Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate 2006, 66, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, B.; Chen, X.; Su, L.; Pu, W.; Wu, J.; Zhu, Z. PTP1B expression contributes to gastric cancer progression. Med. Oncol. 2012, 29, 948–956. [Google Scholar] [CrossRef]
- Wang, X.-M.; Shang, L.; Zhang, Y.; Hao, J.-J.; Shi, F.; Luo, W.; Zhang, T.-T.; Wang, B.-S.; Yang, Y.; Liu, Z.-H.; et al. PTP1B Contributes to Calreticulin-Induced Metastatic Phenotypes in Esophageal Squamous Cell Carcinoma. Mol. Cancer Res. 2013, 11, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Dipikaa Akshinthala, S.; Kragelj, J.; Ringkjøbing Jensen, M.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Dubé, N.; Cheng, A.; Tremblay, M.L. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 1834–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, P.; Eberl, H.C.; Renkawitz, J.; Oslen, J.V.; Tremblay, M.L.; Mann, M.; Ullrich, A.; Daub, H. Investigation of Protein-tyrosine Phosphatase 1B Function by Quantitative Proteomics. Mol. Cell. Proteom. 2008, 7, 1763–1777. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Kim, M.; Baek, M.; Morales, L.D.; Jang, I.-S.; Slaga, T.; DiGiovanni, J.; Kim, D.J. Targeted disruption of TC-PTP in the proliferative compartment augments STAT3 and AKT signaling and skin tumor development. Sci. Rep. 2017, 7, 45077. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.; Lerner, J.; et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Ling, Z.Q.; Zhao, T.; Fang, S.H.; Chang, W.-C.; Lee, S.-C.; Lee, K.-R. Genomic-wide analysis of lymphatic metastasis-associated genes in human hepatocellular carcinoma. World J. Gastroenterol. 2009, 15, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.J.; Hauser, C.; Bukczynska, P.E.; Court, N.W.; Tiganis, T. DNA Replication Stalling Attenuates Tyrosine Kinase Signaling to Suppress S Phase Progression. Cancer Cell 2008, 14, 166–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, B.J.; Tiganis, T. Replication checkpoint control by a PTK/STAT3/Cyclin D1 axis. Cell Cycle 2009, 8, 223–230. [Google Scholar] [CrossRef]
- Kon, S.; Fujita, Y. Cell competition-induced apical elimination of transformed cells, EDAC, orchestrates the cellular homeostasis. Dev. Biol. 2021, 476, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Norman, M. Loss of Scribble causes cell competition in mammalian cells. J. Cell Sci. 2012, 125, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Tamori, Y.; Bialucha, C.; Tian, A.-G.; Kajita, M.; Huan, Y.-C.; Norman, M.; Harrison, N.; Poulton, J.; Ivanovitch, K.; Disch, L.; et al. Involvement of Lgl and Mahjong/VprBP in Cell Competition. PLoS Biol. 2010, 8, 1000422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, L.M.; Montalbano, J.A.; Mihai, C.; Macara, I.G. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 2012, 22, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Stephens, R.; Lim, K.; Portela, M.; Kvansakul, M.; Humbert, P.O.; Richardson, H.E. The Scribble Cell Polarity Module in the Regulation of Cell Signaling in Tissue Development and Tumorigenesis. J. Mol. Biol. 2018, 430, 3585–3612. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.-S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous Activation of Ras and Jak/Stat Pathways in Human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Dong, Y.-L. Cooperation between oncogenic Ras and wild-type p53 stimulates STAT non-cell autonomously to promote tumor radioresistance. Commun. Biol. 2021, 4, 374. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Mancera-Perez, P.A.; Dow, L.E.; Ryan, A.; Tennstedt, P.; Bogani, D.; Elsum, I.; Greenfield, A.; Tuveson, D.A.; Simon, R.; et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J. Clin. Investig. 2011, 121, 4257–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gödde, N.J.; Pearson, H.B.; Smith, L.K.; Humbert, P.O. Dissecting the role of polarity regulators in cancer through the use of mouse models. Exp. Cell Res. 2014, 328, 249–257. [Google Scholar] [CrossRef]
- Lee, M.; Vasioukhin, V. Cell polarity and cancer—Cell and tissue polarity as a non-canonical tumor suppressor. J. Cell Sci. 2008, 121, 1141–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthuswamy, S.K.; Xue, B. Cell Polarity as a Regulator of Cancer Cell Behavior Plasticity. Annu. Rev. Cell Dev. Biol. 2012, 28, 599–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royer, C.; Lu, X. Epithelial cell polarity: A major gatekeeper against cancer? Cell Death Differ. 2011, 18, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- La Marca, J.E.; Diepstraten, S.T.; Hodge, A.L.; Wang, H.; Hart, A.H.; Richardson, H.E.; Somers, W.G. Strip and Cka negatively regulate JNK signalling during Drosophila spermatogenesis. Development 2019, 146, dev174292. [Google Scholar] [CrossRef] [Green Version]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.-C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Marca, J.E.; Willoughby, L.F.; Allan, K.; Portela, M.; Goh, P.K.; Tiganis, T.; Richardson, H.E. PTP61F Mediates Cell Competition and Mitigates Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 12732. https://doi.org/10.3390/ijms222312732
La Marca JE, Willoughby LF, Allan K, Portela M, Goh PK, Tiganis T, Richardson HE. PTP61F Mediates Cell Competition and Mitigates Tumorigenesis. International Journal of Molecular Sciences. 2021; 22(23):12732. https://doi.org/10.3390/ijms222312732
Chicago/Turabian StyleLa Marca, John E., Lee F. Willoughby, Kirsten Allan, Marta Portela, Pei Kee Goh, Tony Tiganis, and Helena E. Richardson. 2021. "PTP61F Mediates Cell Competition and Mitigates Tumorigenesis" International Journal of Molecular Sciences 22, no. 23: 12732. https://doi.org/10.3390/ijms222312732
APA StyleLa Marca, J. E., Willoughby, L. F., Allan, K., Portela, M., Goh, P. K., Tiganis, T., & Richardson, H. E. (2021). PTP61F Mediates Cell Competition and Mitigates Tumorigenesis. International Journal of Molecular Sciences, 22(23), 12732. https://doi.org/10.3390/ijms222312732