
Phospholipase A1 Member A Activates Fibroblast-like Synoviocytes through the Autotaxin-Lysophosphatidic Acid Receptor Axis

 ,
, 

Abstract
:1. Introduction
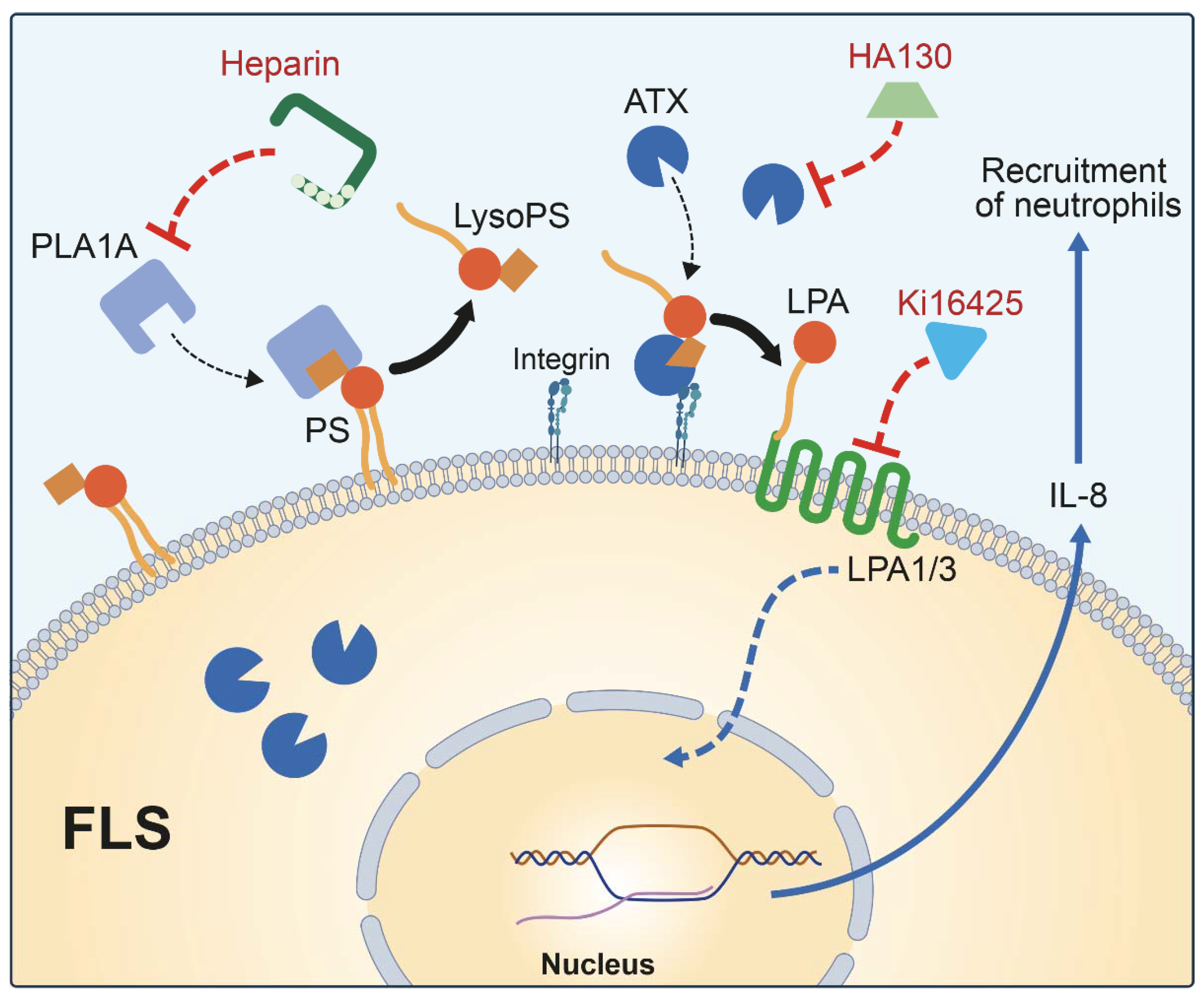
2. Results
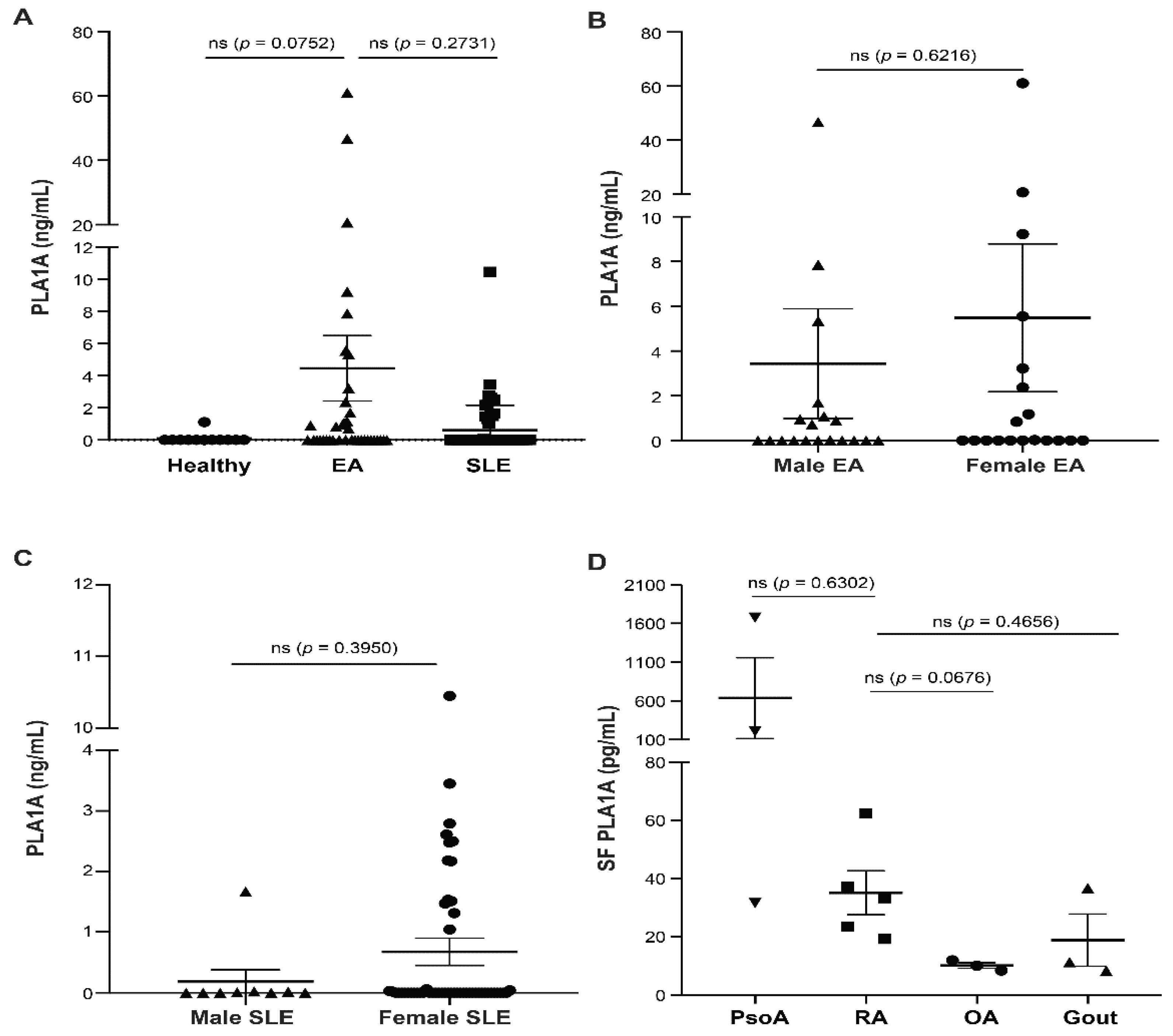
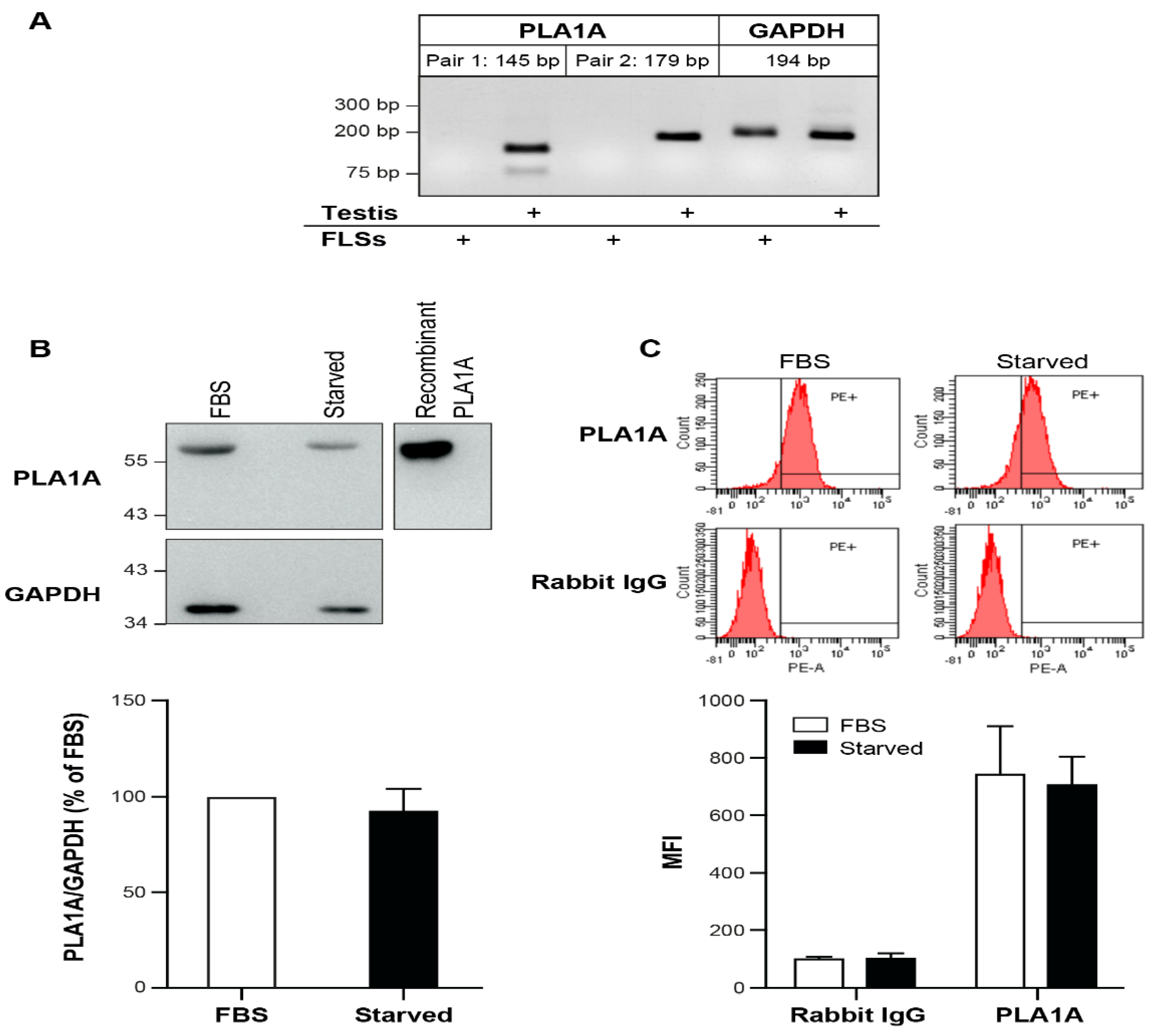
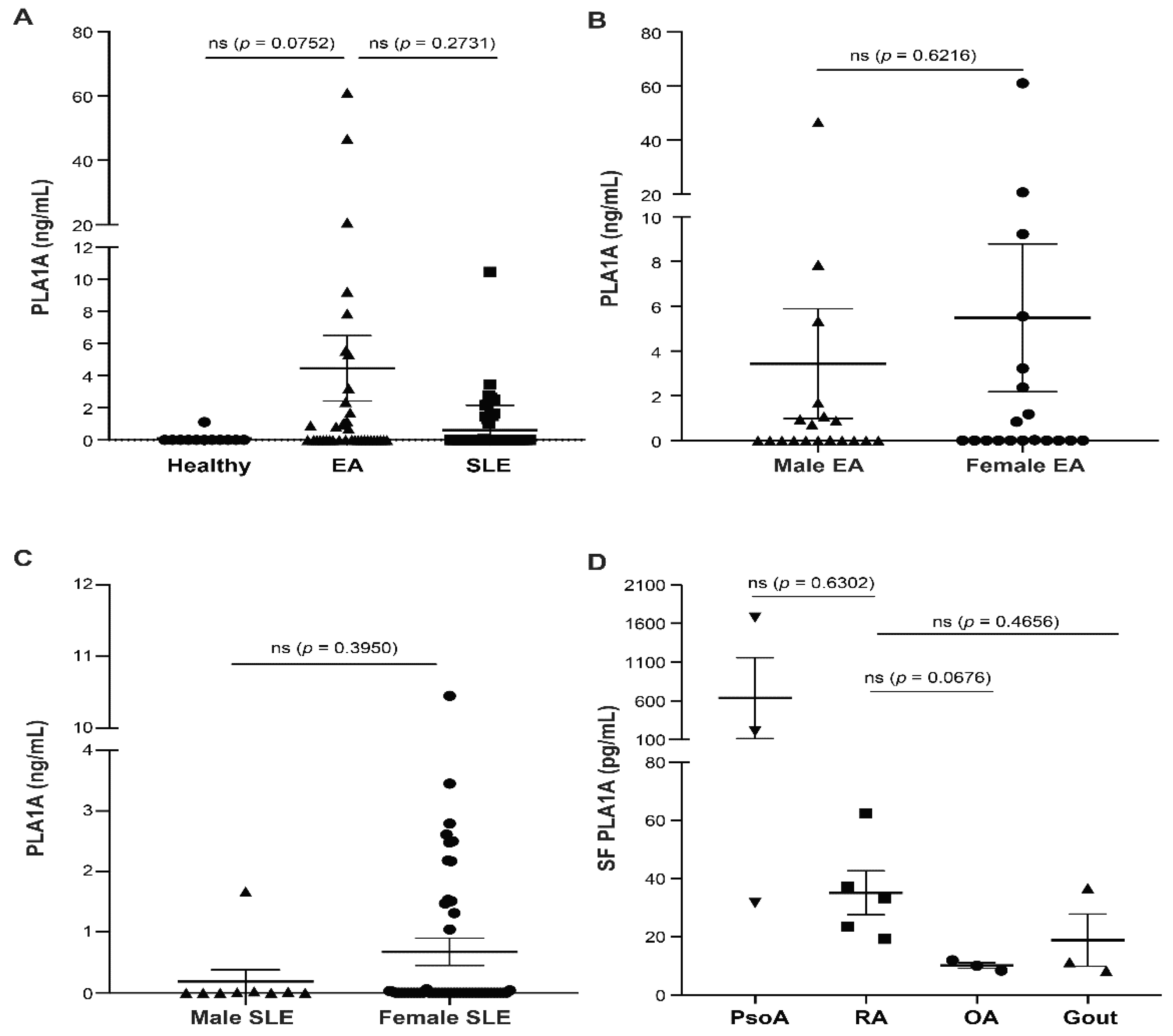
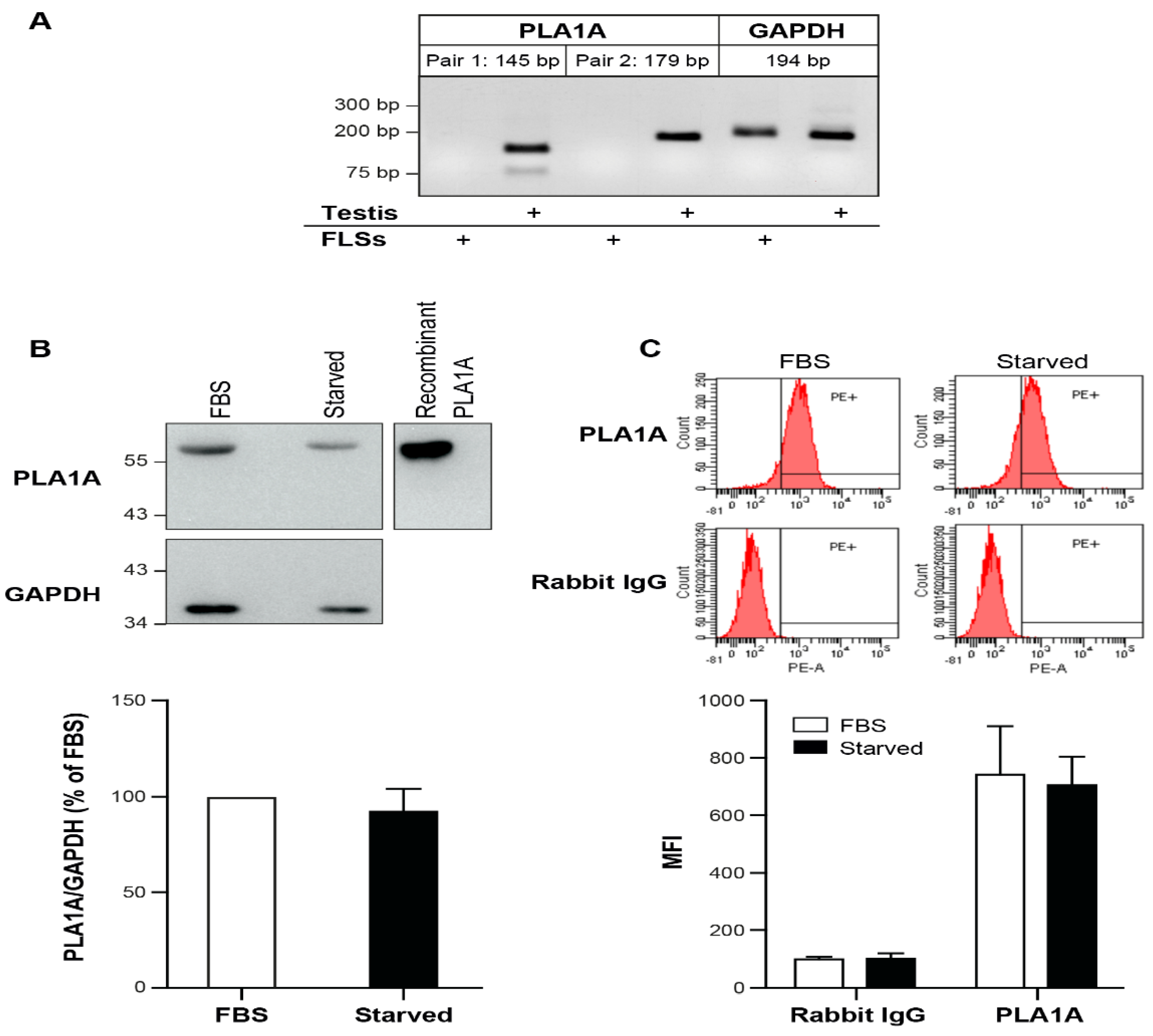
2.1. PLA1A Levels in Plasma, Synovial Fluids, and Human Primary FLSs
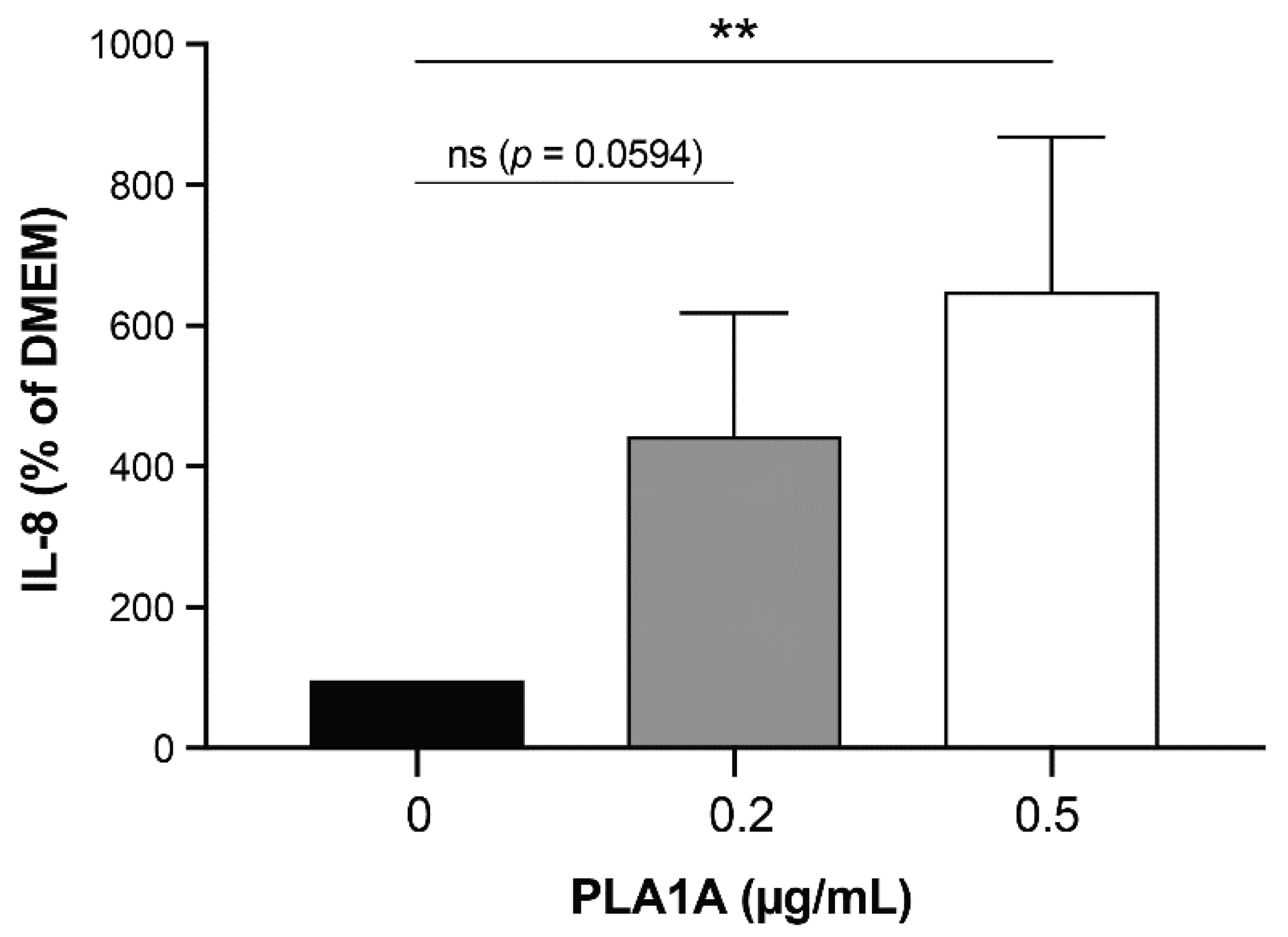
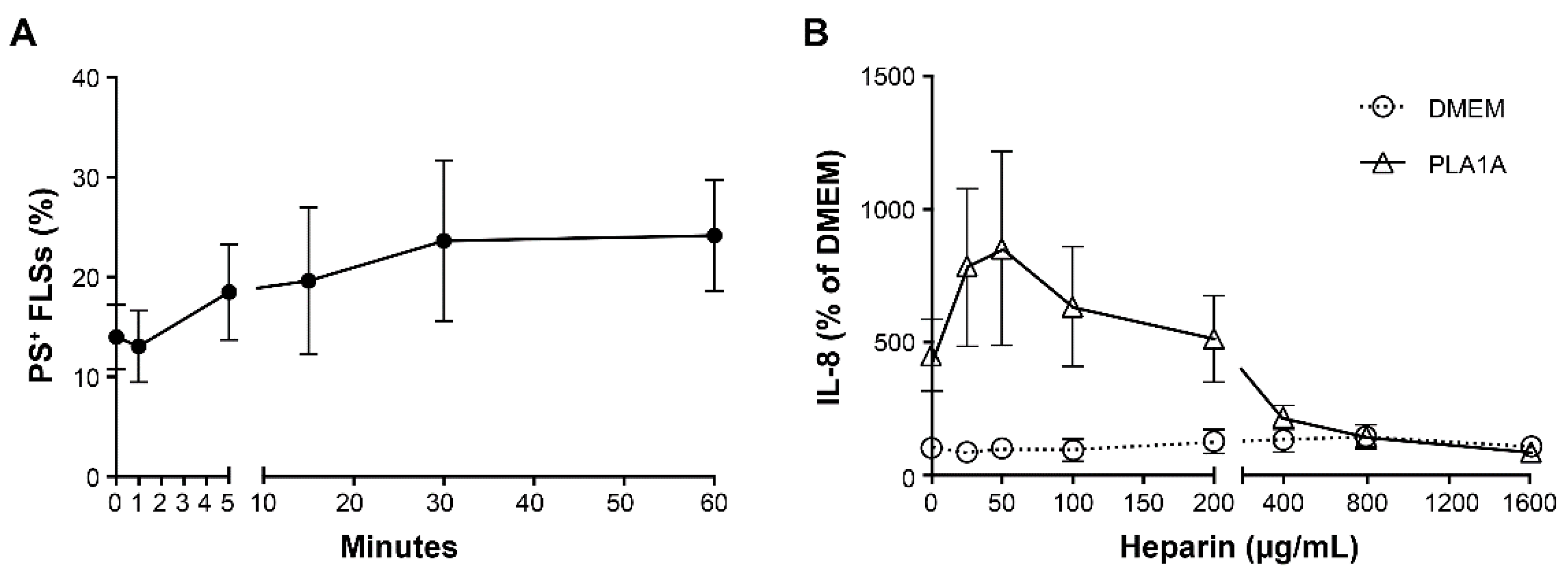
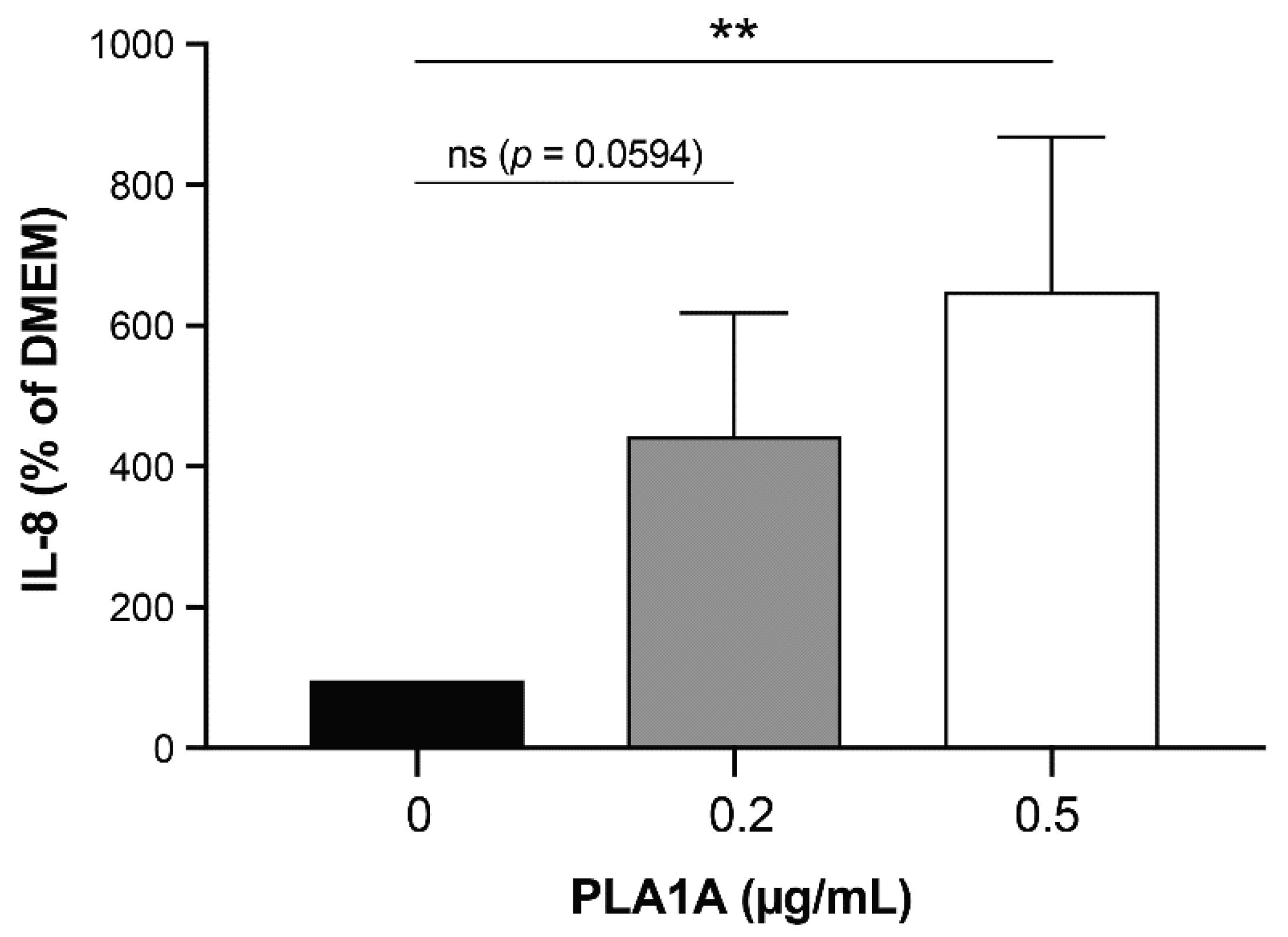
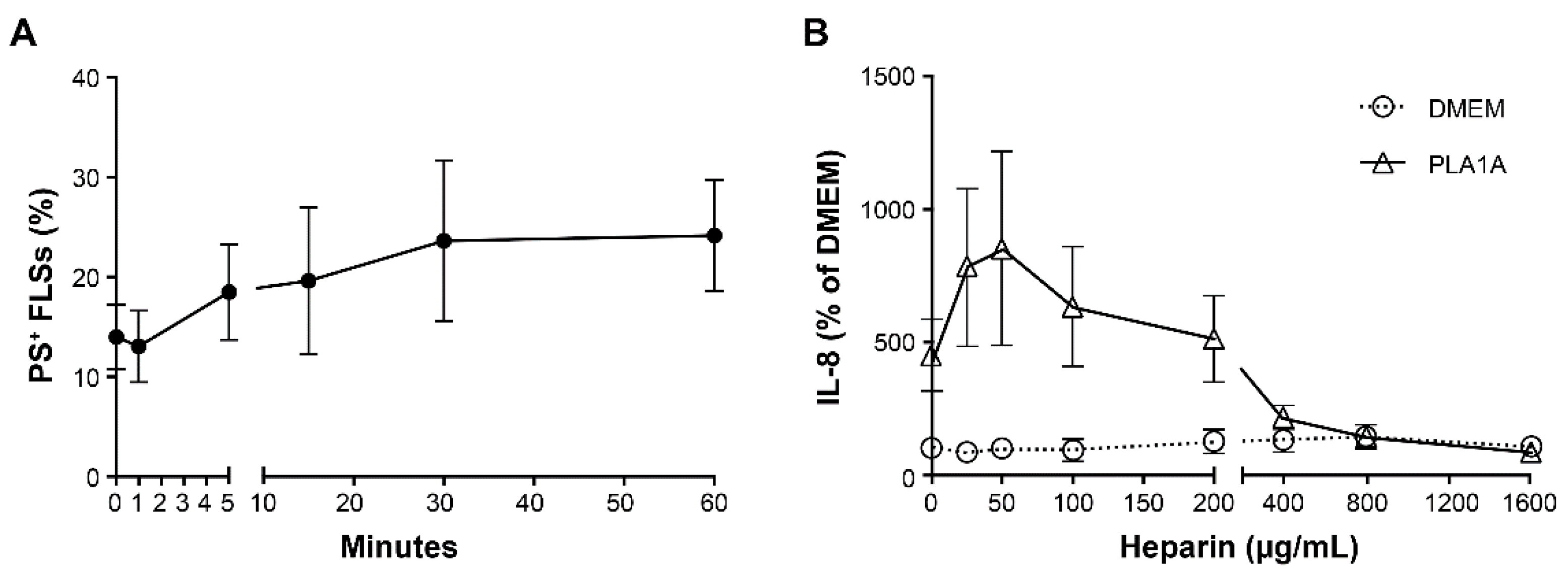
2.2. Pro-Inflammatory Effects of PLA1A and the Detection of Its Substrate
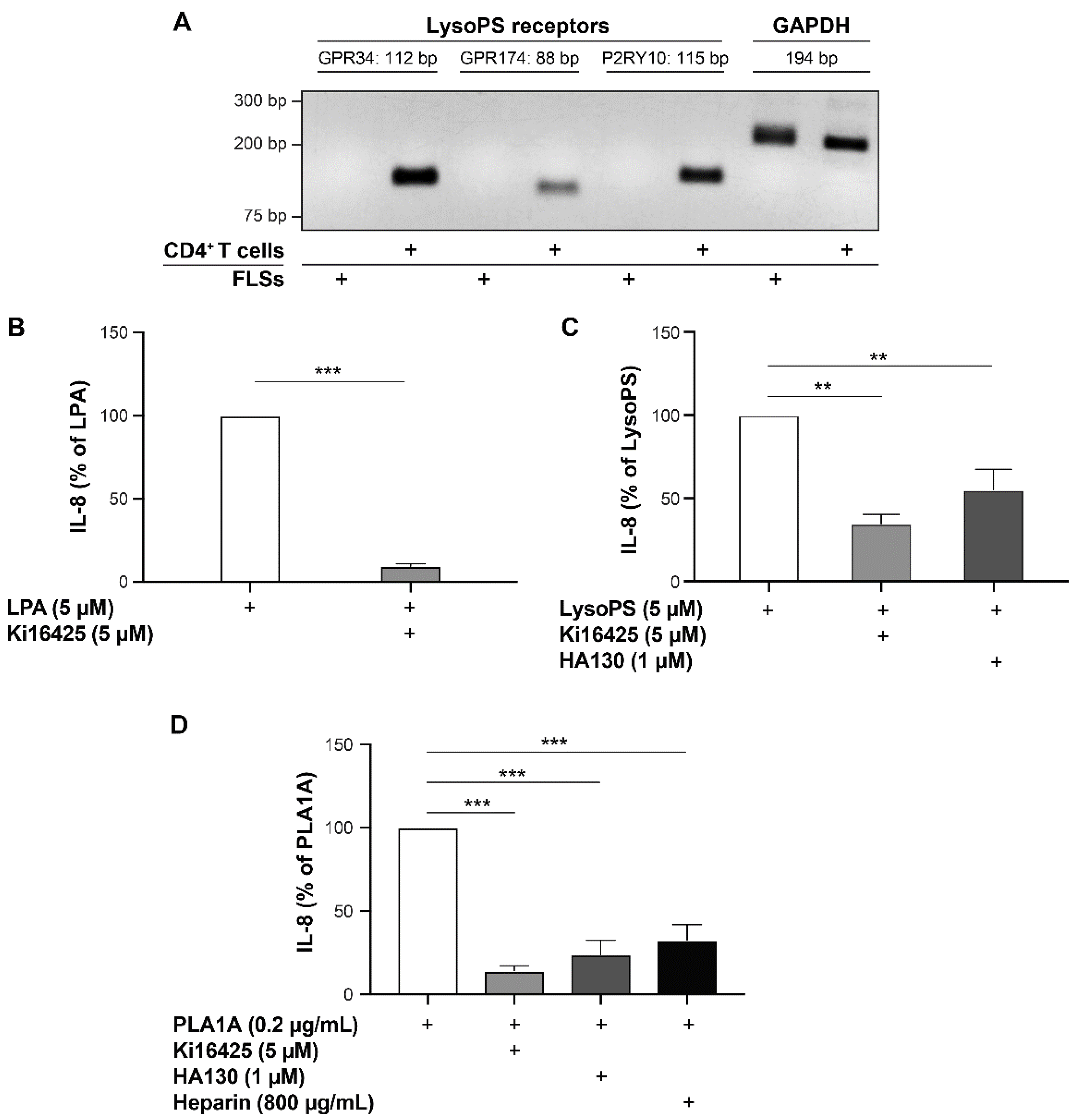
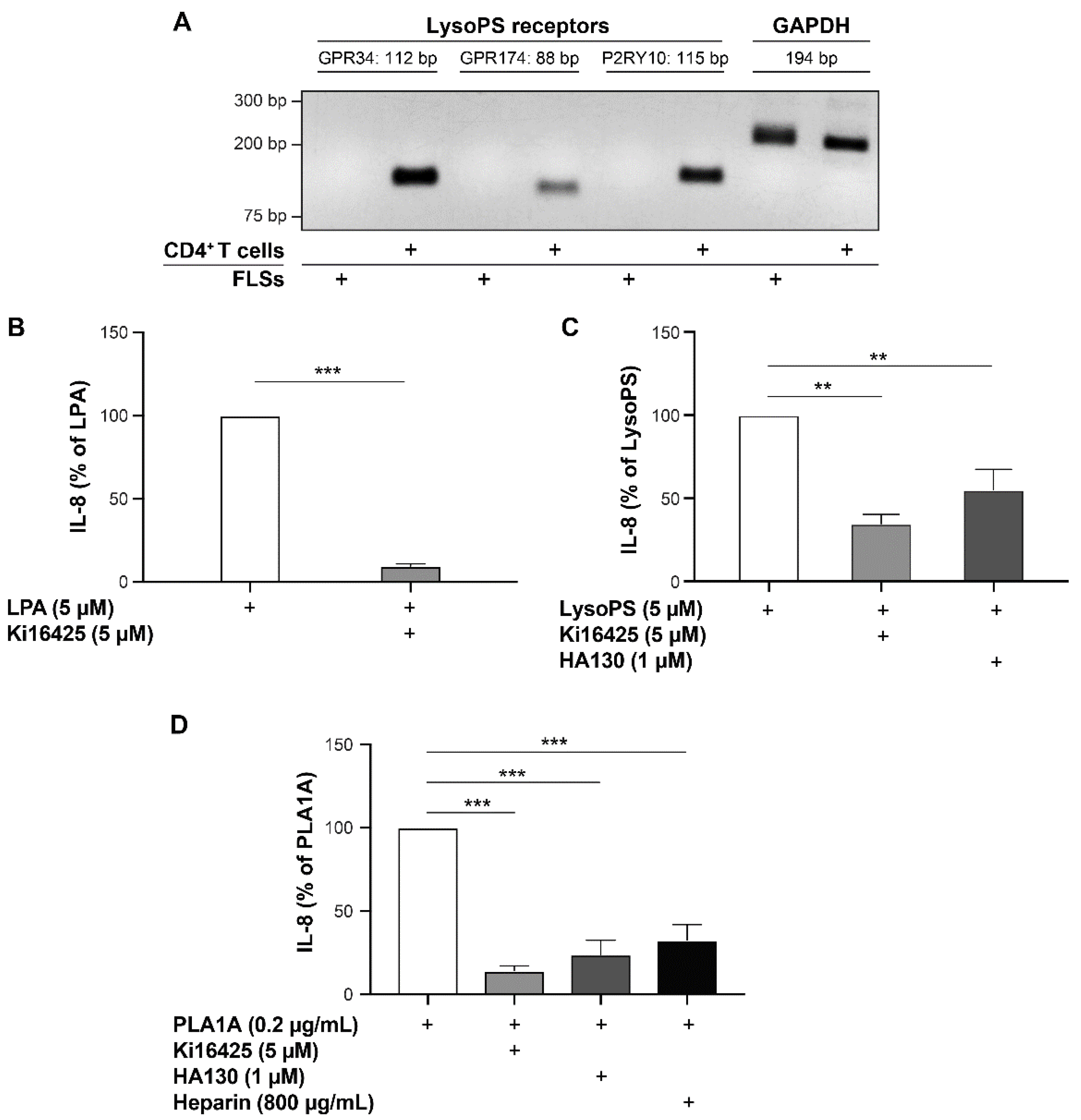
2.3. Pro-Inflammatory Effects of LysoPS and PLA1A in FLSs
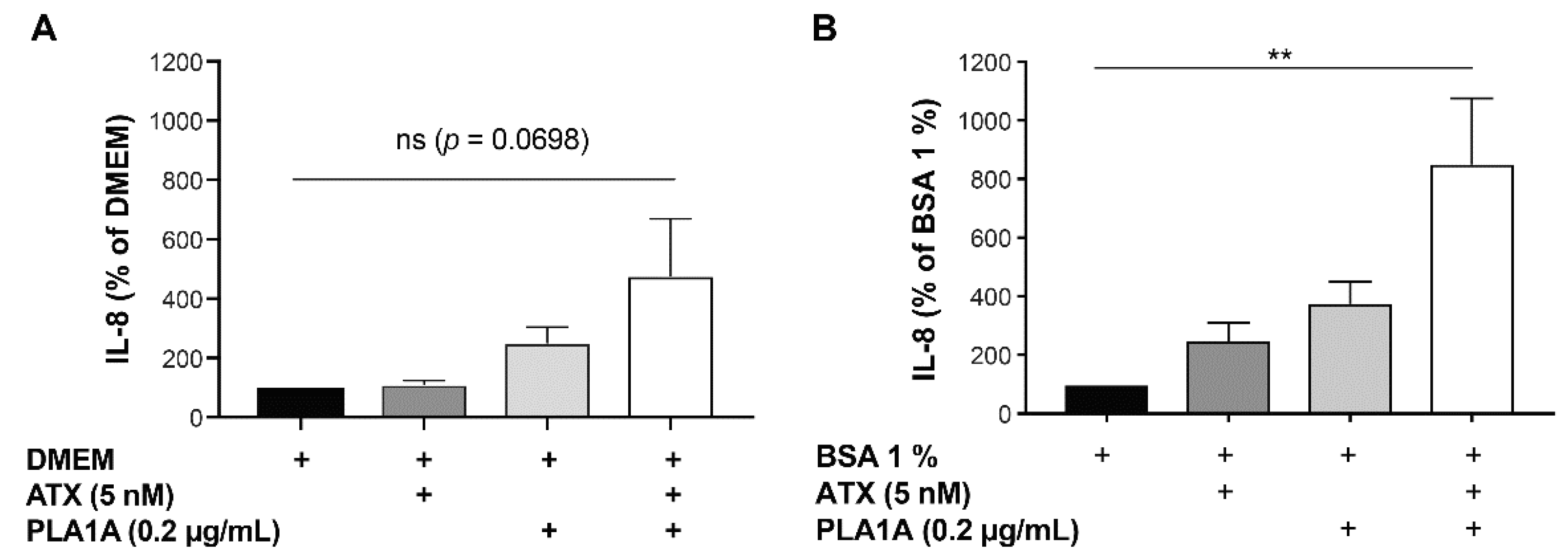
2.4. Effects of PLA1A and ATX in the Presence of Albumin
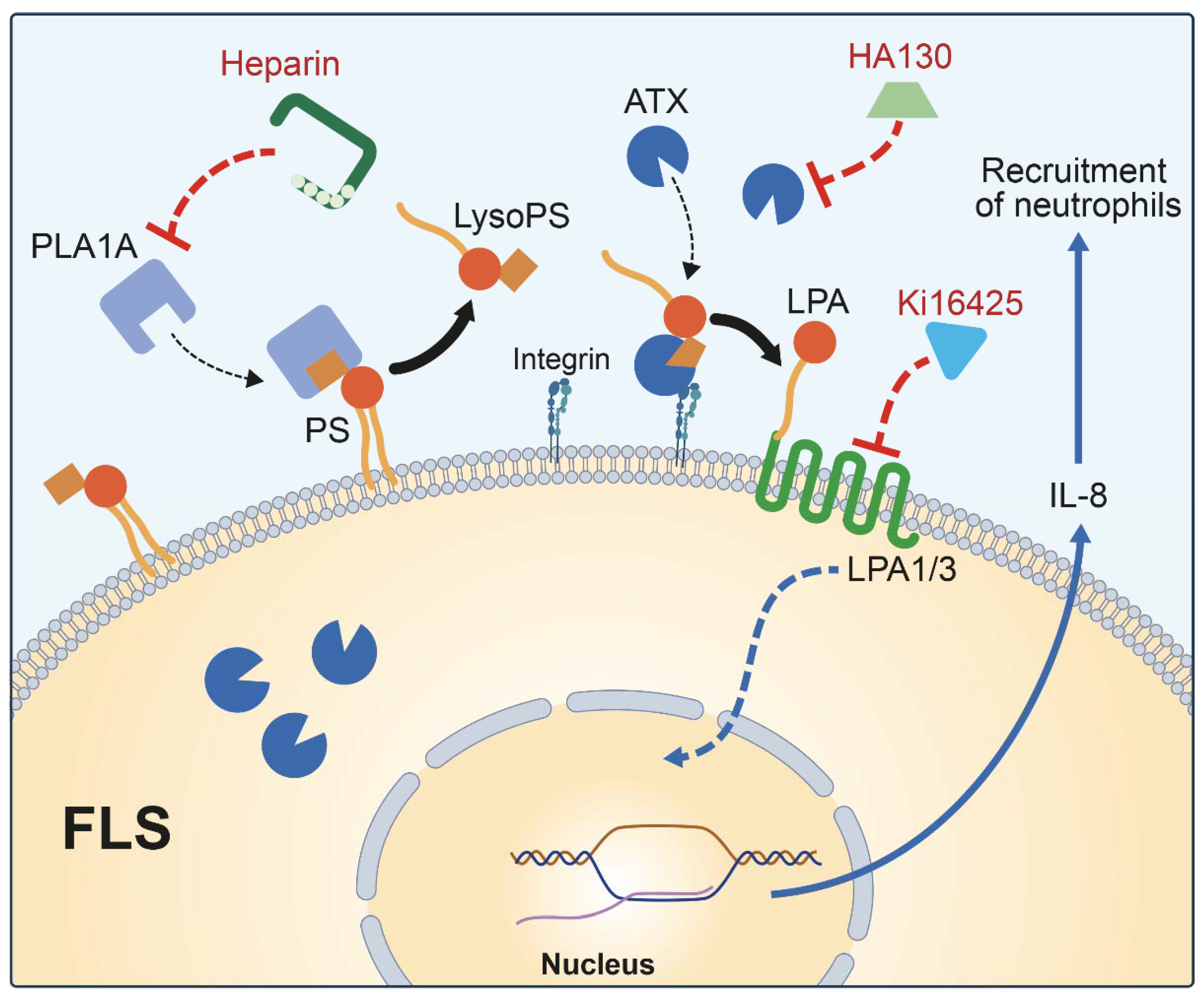
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Human Plasma and Synovial Fluid Samples
4.3. Cell Culture
4.4. Cell Treatment and Viability
4.5. ELISA
4.6. Flow Cytometry
4.7. Semi-Quantitative RT-PCR
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sato, T.; Aoki, J.; Nagai, Y.; Dohmae, N.; Takio, K.; Doi, T.; Arai, H.; Inoue, K. Serine Phospholipid-specific Phospholipase A That Is Secreted from Activated Platelets. J. Biol. Chem. 1997, 272, 2192–2198. [Google Scholar] [CrossRef] [Green Version]
- Aoki, J.; Nagai, Y.; Hosono, H.; Inoue, K.; Arai, H. Structure and function of phosphatidylserine-specific phospholipase A1. Biochim. Biophys. Acta (BBA)-Bioenerg. 2002, 1582, 26–32. [Google Scholar] [CrossRef]
- Zhao, Y.; Hasse, S.; Bourgoin, S.G. Phosphatidylserine-specific phospholipase A1: A friend or the devil in disguise. Prog. Lipid Res. 2021, 83, 101112. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, S.J.; Park, G.Y.; Christman, J.W.; Nyenhuis, S.; Berdyshev, E.; Natarajan, V. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark. Med. 2016, 10, 123–135. [Google Scholar] [CrossRef]
- Loupy, A.; Van Huyen, J.P.D.; Hidalgo, L.; Reeve, J.; Racapé, M.; Aubert, O.; Venner, J.M.; Falmuski, K.; Bories, M.C.; Beuscart, T.; et al. Gene Expression Profiling for the Identification and Classification of Antibody-Mediated Heart Rejection. Circulation 2017, 135, 917–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Groningen, J.J.; Egmond, M.R.; Bloemers, H.P.; Swart, G.W. nmd, a novel gene differentially expressed in human melanoma cell lines, encodes a new atypical member of the enzyme family of lipases. FEBS Lett. 1997, 404, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.-J.; Fraser, M.; Van der Kwast, T.; Boutros, P.C.; Bristow, R.; et al. TMPRSS2–ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Sawada, T.; Kurano, M.; Shirai, H.; Iwasaki, Y.; Tahara, K.; Hayashi, H.; Igarashi, K.; Fujio, K.; Aoki, J.; Yatomi, Y. Serum phosphatidylserine-specific phospholipase A 1 as a novel biomarker for monitoring systemic lupus erythematosus disease activity. Int. J. Rheum. Dis. 2019, 22, 2059–2066. [Google Scholar] [CrossRef]
- Iida, Y.; Sunami, E.; Yamashita, H.; Hiyoshi, M.; Ishihara, S.; Yamaguchi, H.; Inoue, A.; Makide, K.; Tsuno, N.H.; Aoki, J.; et al. Phosphatidylserine-specific phospholipase A1 (PS-PLA1) expression in colorectal cancer correlates with tumor invasion and hematogenous metastasis. Anticancer Res. 2015, 35, 1459–1464. [Google Scholar]
- Kurano, M.; Miyagaki, T.; Miyagawa, T.; Igarashi, K.; Shimamoto, S.; Ikeda, H.; Aoki, J.; Sato, S.; Yatomi, Y. Association between serum autotaxin or phosphatidylserine-specific phospholipase A1 levels and melanoma. J. Dermatol. 2018, 45, 571–579. [Google Scholar] [CrossRef]
- Nakawatari, K.; Kurano, M.; Araki, O.; Nishikawa, M.; Shimamoto, S.; Igarashi, K.; Aoki, J.; Murakami, M.; Yatomi, Y. Elevated phosphatidylserine-specific phospholipase A1 level in hyperthyroidism. Clin. Chim. Acta 2020, 503, 99–106. [Google Scholar] [CrossRef]
- Kurano, M.; Dohi, T.; Nojiri, T.; Kobayashi, T.; Hirowatari, Y.; Inoue, A.; Kano, K.; Matsumoto, H.; Igarashi, K.; Nishikawa, M.; et al. Blood levels of serotonin are specifically correlated with plasma lysophosphatidylserine among the glycero-lysophospholipids. BBA Clin. 2015, 4, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Kurano, M.; Kano, K.; Dohi, T.; Matsumoto, H.; Igarashi, K.; Nishikawa, M.; Ohkawa, R.; Ikeda, H.; Miyauchi, K.; Daida, H.; et al. Different origins of lysophospholipid mediators between coronary and peripheral arteries in acute coronary syndrome. J. Lipid Res. 2017, 58, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Emoto, S.; Kurano, M.; Kano, K.; Matsusaki, K.; Yamashita, H.; Nishikawa, M.; Igarashi, K.; Ikeda, H.; Aoki, J.; Kitayama, J.; et al. Analysis of glycero-lysophospholipids in gastric cancerous ascites. J. Lipid Res. 2017, 58, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makide, K.; Uwamizu, A.; Shinjo, Y.; Ishiguro, J.; Okutani, M.; Inoue, A.; Aoki, J. Novel lysophosphoplipid receptors: Their structure and function. J. Lipid Res. 2014, 55, 1986–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugo, T.; Tachimoto, H.; Chikatsu, T.; Murakami, Y.; Kikukawa, Y.; Sato, S.; Kikuchi, K.; Nagi, T.; Harada, M.; Ogi, K.; et al. Identification of a lysophosphatidylserine receptor on mast cells. Biochem. Biophys. Res. Commun. 2006, 341, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.M.; Kim, H.J.; Kim, S.M.; Jung, Y.; Park, S.W.; Chung, I.Y. Lysophosphatidylserine receptor P2Y10: A G protein-coupled receptor that mediates eosinophil degranulation. Clin. Exp. Allergy 2018, 48, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Frasch, S.C.; Fernandez-Boyanapalli, R.F.; Berry, K.A.Z.; Murphy, R.C.; Leslie, C.C.; Nick, J.A.; Henson, P.M.; Bratton, D.L. Neutrophils Regulate Tissue Neutrophilia in Inflammation via the Oxidant-modified Lipid Lysophosphatidylserine. J. Biol. Chem. 2013, 288, 4583–4593. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.J.; Li, C.-M.; Xu, Y.; An, J.; Huang, Y.; Cyster, J.G. The lysophosphatidylserine receptor GPR174 constrains regulatory T cell development and function. J. Exp. Med. 2015, 212, 1011–1020. [Google Scholar] [CrossRef]
- Nishikawa, M.; Kurano, M.; Ikeda, H.; Aoki, J.; Yatomi, Y. Lysophosphatidylserine has Bilateral Effects on Macrophages in the Pathogenesis of Atherosclerosis. J. Atheroscler. Thromb. 2015, 22, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Frasch, S.C.; Bratton, D.L. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog. Lipid Res. 2012, 51, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; A Liotta, L. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [CrossRef]
- Pamuklar, Z.; Federico, L.; Liu, S.; Umezu-Goto, M.; Dong, A.; Panchatcharam, M.; Fulerson, Z.; Berdyshev, E.; Natarajan, V.; Fang, X.; et al. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J. Biol. Chem. 2009, 284, 7385–7394. [Google Scholar] [CrossRef] [Green Version]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2008, 1781, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum Lysophosphatidic Acid Is Produced through Diverse Phospholipase Pathways. J. Biol. Chem. 2002, 277, 48737–48744. [Google Scholar] [CrossRef] [Green Version]
- van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The Autotaxin–LPA Axis Emerges as a Novel Regulator of Lymphocyte Homing and Inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Rives, S.A.; González-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef]
- Lin, Y.H.; Lin, Y.C.; Chen, C.C. Lysophosphatidic Acid Receptor Antagonists and Cancer: The Current Trends, Clinical Implications, and Trials. Cells 2021, 10, 1629. [Google Scholar] [CrossRef]
- Wu, P.-Y.; Lin, Y.-C.; Huang, Y.-L.; Chen, W.-M.; Chen, C.-C.; Lee, H. Mechanisms of Lysophosphatidic Acid-Mediated Lymphangiogenesis in Prostate Cancer. Cancers 2018, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hasse, S.; Zhao, C.; Bourgoin, S.G. Targeting the autotaxin—Lysophosphatidic acid receptor axis in cardiovascular diseases. Biochem. Pharmacol. 2019, 164, 74–81. [Google Scholar] [CrossRef]
- Bolen, A.L.; Naren, A.P.; Yarlagadda, S.; Beranova-Giorgianni, S.; Chen, L.; Norman, D.; Baker, D.L.; Rowland, M.M.; Best, M.D.; Sano, T.; et al. The phospholipase A1 activity of lysophospholipase A-I links platelet activation to LPA production during blood coagulation. J. Lipid Res. 2011, 52, 958–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandoh, K.; Aoki, J.; Taira, A.; Tsujimoto, M.; Arai, H.; Inoue, K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. FEBS Lett. 2000, 478, 159–165. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosono, H.; Aoki, J.; Nagai, Y.; Bandoh, K.; Ishida, M.; Taguchi, R.; Arai, H.; Inoue, K. Phosphatidylserine-specific Phospholipase A1Stimulates Histamine Release from Rat Peritoneal Mast Cells through Production of 2-Acyl-1-lysophosphatidylserine. J. Biol. Chem. 2001, 276, 29664–29670. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Fernandes, M.J.; Prestwich, G.D.; Turgeon, M.; Di Battista, J.; Clair, T.; Poubelle, P.E.; Bourgoin, S.G. Regulation of Lysophosphatidic Acid Receptor Expression and Function in Human Synoviocytes: Implications for Rheumatoid Arthritis? Mol. Pharmacol. 2008, 73, 587–600. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Igarashi, K.; Ohkawa, R.; Saiki, N.; Nagasaki, M.; Uno, K.; Hayashi, N.; Sawada, T.; Syukuya, K.; Yokota, H.; et al. A novel enzyme immunoassay for the determination of phosphatidylserine-specific phospholipase A1 in human serum samples. Clin. Chim. Acta 2010, 411, 1090–1094. [Google Scholar] [CrossRef]
- Nagai, Y.; Aoki, J.; Sato, T.; Amano, K.; Matsuda, Y.; Arai, H.; Inoue, K. An Alternative Splicing Form of Phosphatidylserine-specific Phospholipase A1 That Exhibits Lysophosphatidylserine-specific Lysophospholipase Activity in Humans. J. Biol. Chem. 1999, 274, 11053–11059. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clézardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood J. Am. Soc. Hematol. 2014, 124, 3141–3150. [Google Scholar]
- Aoki, J.; Inoue, A.; Makide, K.; Saiki, N.; Arai, H. Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie 2007, 89, 197–204. [Google Scholar] [CrossRef]
- Uranbileg, B.; Kurano, M.; Sato, M.; Ikeda, H.; Ishizawa, T.; Hasegawa, K.; Kokudo, N.; Yatomi, Y. Possible involvement of PS-PLA1 and lysophosphatidylserine receptor (LPS1) in hepatocellular carcinoma. Sci. Rep. 2020, 10, 2659. [Google Scholar] [CrossRef] [Green Version]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef]
- Koch, A.E. Chemokines and their receptors in rheumatoid arthritis: Future targets? Arthritis Rheum. 2005, 52, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; He, Y.; Wang, B.; Xun, Z.; Chen, S.; Zeng, Z.; Ou, Q. Blocking of YY1 reduce neutrophil infiltration by inhibiting IL-8 production via the PI3K-Akt-mTOR signaling pathway in rheumatoid arthritis. Clin. Exp. Immunol. 2019, 195, 226–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, L.A.; Lubberts, E.; Helsen, M.M.; Saxne, T.; Coenen-de Roo, C.J.; Heinegård, D.; van den Berg, W.B. Protection against cartilage and bone destruction by systemic interleukin-4 treatment in established murine type II collagen-induced arthritis. Arthritis Res. 1999, 1, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiramatsu, T.; Sonoda, H.; Takanezawa, Y.; Morikawa, R.; Ishida, M.; Kasahara, K.; Sanai, Y.; Taguchi, R.; Aoki, J.; Arai, H. Biochemical and Molecular Characterization of Two Phosphatidic Acid-selective Phospholipase A1s, mPA-PLA1α and mPA-PLA1β. J. Biol. Chem. 2003, 278, 49438–49447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Okada, Y.; Saito, K.; Tanaka, Y. Induction of RANKL expression and osteoclast maturation by the binding of fibroblast growth factor 2 to heparan sulfate proteoglycan on rheumatoid synovial fibroblasts. Arthritis Rheum. 2004, 50, 2450–2458. [Google Scholar] [CrossRef] [PubMed]
- Bourgoin, S.G.; Zhao, C. Autotaxin and lysophospholipids in rheumatoid arthritis. Curr. Opin. Investig. Drugs 2010, 11, 515–526. [Google Scholar]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Jethwa, S.A.; Leah, E.J.; Zhang, Q.; Bright, N.A.; Oxley, D.; Bootman, M.; Rudge, S.A.; Wakelam, M.J.O. Exosomes bind autotaxin and act as a physiological delivery mechanism to stimulate LPA receptor signalling in cells. J. Cell Sci. 2016, 129, 3948–3957. [Google Scholar] [CrossRef] [Green Version]
- Moolenaar, W.H.; Perrakis, A. Insights into autotaxin: How to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 2011, 12, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Polo, F.; Fish, A.; Matsoukas, M.-T.; Heidebrecht, T.; Keune, W.-J.; Perrakis, A. Lysophosphatidic acid produced by autotaxin acts as an allosteric modulator of its catalytic efficiency. J. Biol. Chem. 2018, 293, 14312–14327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of Autotaxin to Integrins Localizes Lysophosphatidic Acid Production to Platelets and Mammalian Cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meeteren, L.A.; Ruurs, P.; Christodoulou, E.; Goding, J.W.; Takakusa, H.; Kikuchi, K.; Perrakis, A.; Nagano, T.; Moolenaar, W.H. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005, 280, 21155–21161. [Google Scholar] [CrossRef] [Green Version]
- Benesch, M.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Tokumura, A.; Iimori, M.; Nishioka, Y.; Kitahara, M.; Sakashita, M.; Tanaka, S. Lysophosphatidic acids induce proliferation of cultured vascular smooth muscle cells from rat aorta. Am. J. Physiol. Physiol. 1994, 267, C204–C210. [Google Scholar] [CrossRef]
- Hayashi, K.; Takahashi, M.; Nishida, W.; Yoshida, K.; Ohkawa, Y.; Kitabatake, A.; Aoki, J.; Arai, H.; Sobue, K. Phenotypic Modulation of Vascular Smooth Muscle Cells Induced by Unsaturated Lysophosphatidic Acids. Circ. Res. 2001, 89, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sense | Antisense | ||
|---|---|---|---|
| GAPDH | 5′-AAT CCC ATC ACC ATC TTC CAG-3′ | 5′-TTC ACA CCC ATG ACG AAC AT-3′ | |
| PLA1A | Pair 1 | 5′-GCC CAA GGA TAG GAC TGG TG-3′ | 5′-GCG TTG CTG CTT AGG TAT GG-3′ |
| Pair 2 | 5′-CCA TCC ACA CAG ACA CCG AC-3′ | 5′-TGA CAC CAC CTT GTT CCA CC-3′ | |
| GPR34 | Pair 1 | 5′-TGT ATT TCC TGA TGT CCA GTA AT-3′ | 5′-GCT TTC ACT TCT GCT TGC TT-3′ |
| Pair 2 | 5′-GGG ACT GGT TGG GAA CAT AAT-3′ | 5′-GAT GAG TAG GAG GTC TGC AAT G-3′ | |
| Pair 3 | 5′-CAG CAA CGG AAG GCA ATA ACA-3′ | 5′-GCT TCT CCT TTT GCG TTA TGC T-3′ | |
| GPR174 | Pair 1 | 5′-GCC TTA TGG GTA TTT TAC GG-3′ | 5′-AGT CAG CAA TGG CTA GGT TT-3′ |
| Pair 2 | 5′-GGG TTT GTA ACT CCG CTT CT-3′ | 5′-GAT CTT GGG CCA TGG GAT ATT-3′ | |
| Pair 3 | 5′-ATC AGT GTG CGA CGA TTT TGG-3′ | 5′-AAA GAG TAC ACA GGC AAG GCA-3′ | |
| P2RY10 | Pair 1 | 5′-AGT CTT CGT TAT CTG CTT CAC T-3′ | 5′-GAT GGA AAT ACA GGG TAC TTT T-3′ |
| Pair 2 | 5′-CTC TAT GCT GGT CAT TCC CTT C-3′ | 5′-CCA GGG TAC ATG CTT CCT ATT C-3′ | |
| Pair 3 | 5′-TAG CAG TTG TCC CGT TGT CC-3′ | 5′-CAT GGC GGG ATA GTT GGT CA-3′ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Hasse, S.; Vaillancourt, M.; Zhao, C.; Davis, L.; Boilard, E.; Fortin, P.; Di Battista, J.; Poubelle, P.E.; Bourgoin, S.G. Phospholipase A1 Member A Activates Fibroblast-like Synoviocytes through the Autotaxin-Lysophosphatidic Acid Receptor Axis. Int. J. Mol. Sci. 2021, 22, 12685. https://doi.org/10.3390/ijms222312685
Zhao Y, Hasse S, Vaillancourt M, Zhao C, Davis L, Boilard E, Fortin P, Di Battista J, Poubelle PE, Bourgoin SG. Phospholipase A1 Member A Activates Fibroblast-like Synoviocytes through the Autotaxin-Lysophosphatidic Acid Receptor Axis. International Journal of Molecular Sciences. 2021; 22(23):12685. https://doi.org/10.3390/ijms222312685
Chicago/Turabian StyleZhao, Yang, Stephan Hasse, Myriam Vaillancourt, Chenqi Zhao, Lynn Davis, Eric Boilard, Paul Fortin, John Di Battista, Patrice E. Poubelle, and Sylvain G. Bourgoin. 2021. "Phospholipase A1 Member A Activates Fibroblast-like Synoviocytes through the Autotaxin-Lysophosphatidic Acid Receptor Axis" International Journal of Molecular Sciences 22, no. 23: 12685. https://doi.org/10.3390/ijms222312685
APA StyleZhao, Y., Hasse, S., Vaillancourt, M., Zhao, C., Davis, L., Boilard, E., Fortin, P., Di Battista, J., Poubelle, P. E., & Bourgoin, S. G. (2021). Phospholipase A1 Member A Activates Fibroblast-like Synoviocytes through the Autotaxin-Lysophosphatidic Acid Receptor Axis. International Journal of Molecular Sciences, 22(23), 12685. https://doi.org/10.3390/ijms222312685