
ENPP2 Promoter Methylation Correlates with Decreased Gene Expression in Breast Cancer: Implementation as a Liquid Biopsy Biomarker

 ,
,  ,
, 
 and
and 
Abstract
:1. Introduction
2. Results
2.1. In Silico Analysis of ENPP2 Methylation and Expression in BrCa
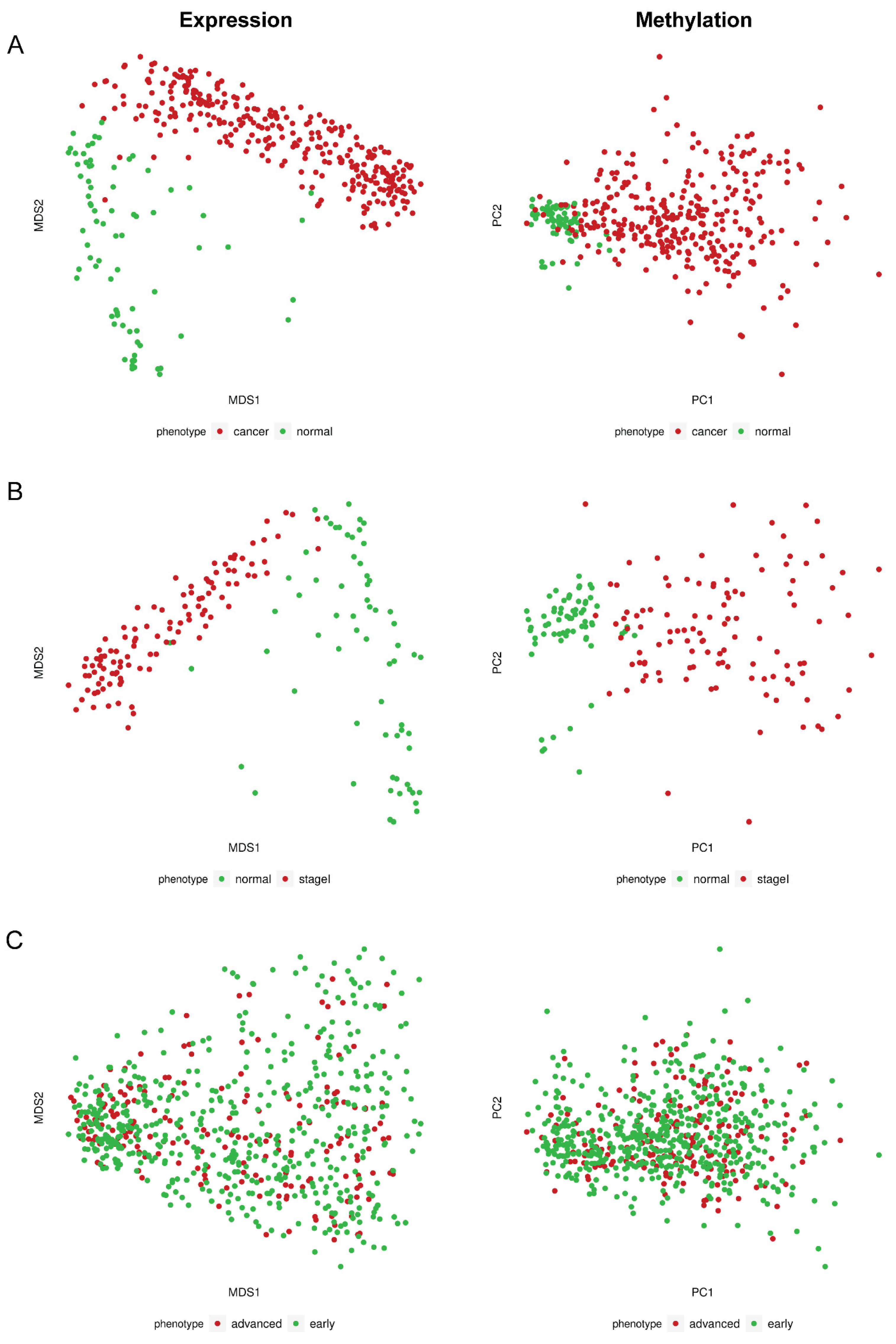
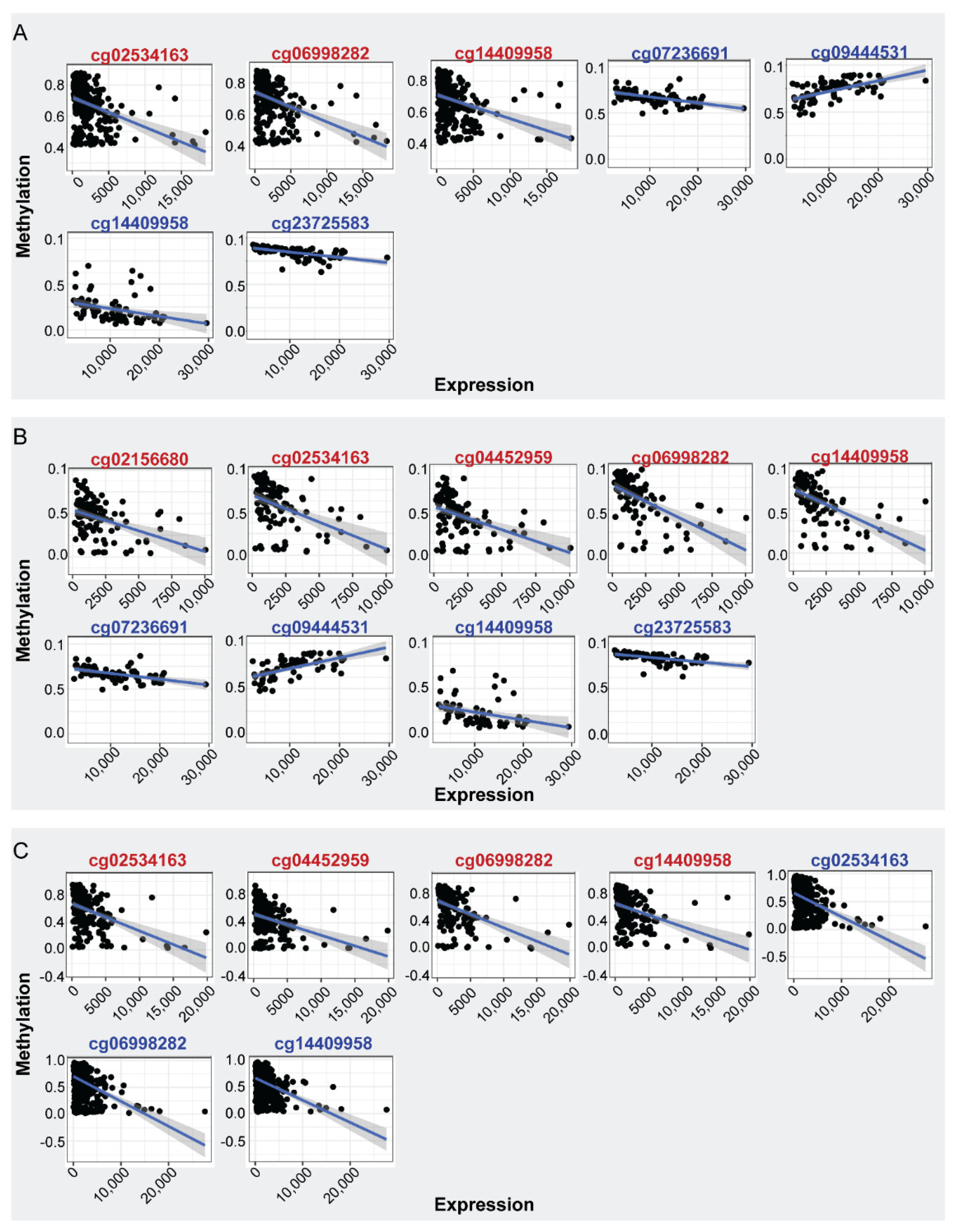
2.1.1. Differential Methylation and Expression Analysis between BrCa and Normal Breast Tissue
2.1.2. Differential Methylation Analysis between Primary and Metastatic BrCa
2.1.3. Differential Methylation and Expression Analysis between Stage I BrCa and Normal
2.1.4. Differential Methylation and Expression Analysis between Early- and Advanced-Stage BrCa
2.1.5. Differential Methylation Analysis between BrCa Cancer Types
2.1.6. In Silico Analysis of ENPP2 Methylation in BrCa ccfDNA Data
2.2. ENPP2 Methylation, Expression and Survival Analysis by UALCAN
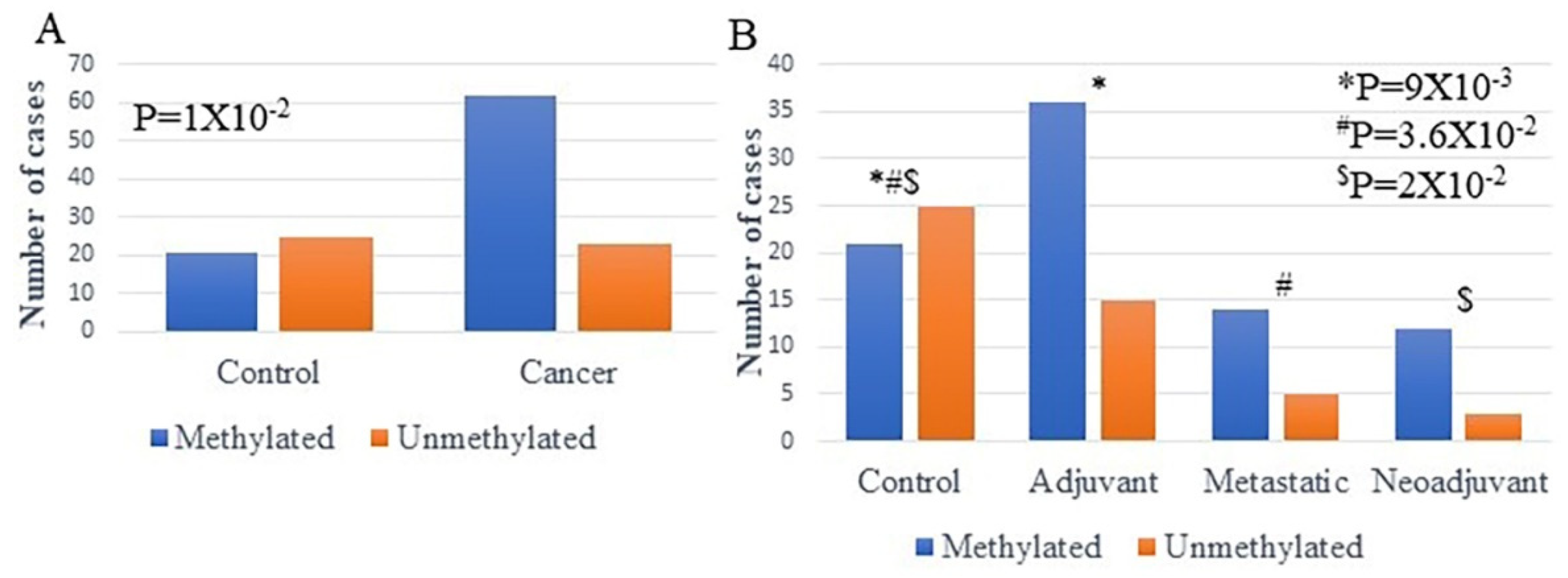
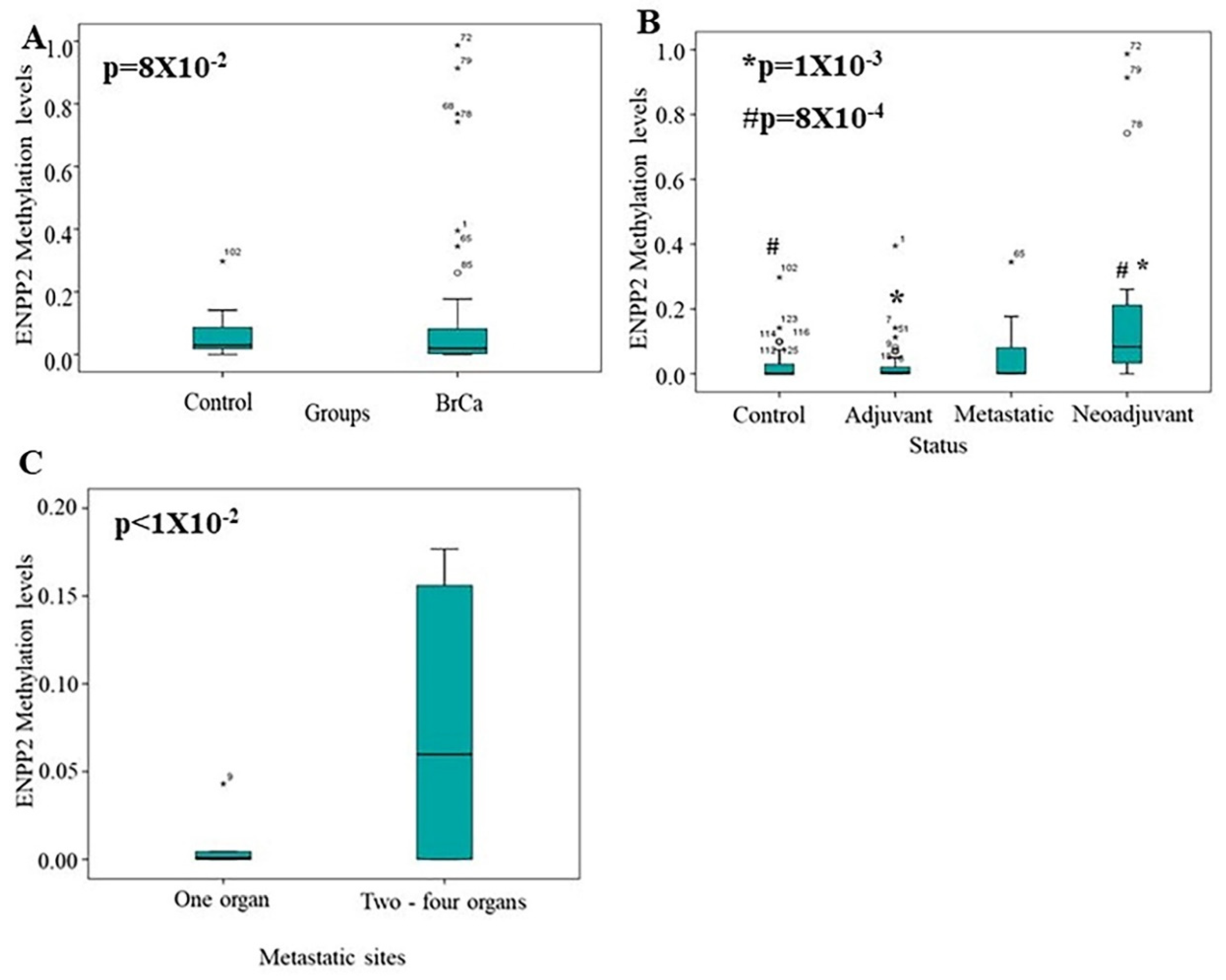
2.3. Methylation Analysis of ENPP2 in ccfDNA from BrCa Patients
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Analysis of ENPP2 in BrCa
4.1.1. Data Sources
4.1.2. Data Preprocessing and DNA Methylation Analysis
4.1.3. Differential Expression Analysis and Expression—Methylation Correlation
4.1.4. Expression, Methylation and Survival Analysis Using the UALCAN Platform
4.2. Methylation Analysis of ENPP2 in BrCa Liquid Biopsies
4.2.1. Study Groups and Clinical Samples
4.2.2. ccfDNA Extraction
4.2.3. Sodium Bisulfite Conversion of ccfDNA
4.2.4. Quantitative Methylation-Specific PCR (qMSP)
4.2.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque-Bolivar, A.; Villegas, V.E.; Rondón-Lagos, M. Resistance and Overcoming Resistance in Breast Cancer. Breast Cancer Dove Med. Press 2020, 12, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.; Valerie, G.B. The role and therapeutic potential of the autotaxin—Lysophosphatidate signalling axis in breast cancer. Biochem. J. 2014, 463, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Brindley, D.N. Autotaxin and Breast Cancer: Towards Overcoming Treatment Barriers and Sequelae. Cancers 2020, 12, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Fang, X.J.; Casey, G.; Mills, G.B. Lysophospholipids activate ovarian and breast cancer cells. Biochem. J. 1995, 309 Pt 3, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Y.; Lee, J.; Park, C.G.; Kim, S.; Hong, S.; Chung, H.C.; Min, S.K.; Han, J.W.; Lee, H.W.; Lee, H.Y. Expression of autotaxin (NPP-2) is closely linked to invasiveness of breast cancer cells. Clin. Exp. Metastasis 2002, 19, 603–608. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Role of the autotaxin–lysophosphatidate axis in the development of resistance to cancer therapy. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158716. [Google Scholar] [CrossRef]
- David, M.; Wannecq, E.; Descotes, F.; Jansen, S.; Deux, B.; Ribeiro, J.; Serre, C.-M.; Grès, S.; Bendriss-Vermare, N.; Bollen, M.; et al. Cancer Cell Expression of Autotaxin Controls Bone Metastasis Formation in Mouse through Lysophosphatidic Acid-Dependent Activation of Osteoclasts. PLoS ONE 2010, 5, e9741. [Google Scholar] [CrossRef]
- Boucharaba, A.; Serre, C.-M.; Grès, S.; Saulnier-Blache, J.S.; Bordet, J.-C.; Guglielmi, J.; Clézardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.H.K.; Maher, E.R. DNA methylation: A form of epigenetic control of gene expression. Obstet. Gynaecol. 2010, 12, 37–42. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Esteller, M.; Chatzaki, E. Circulating Cell-Free DNA in Breast Cancer: Searching for Hidden Information towards Precision Medicine. Cancers 2021, 13, 728. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Cheretaki, A.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Amarantidis, K.; Xenidis, N.; Kakolyris, S.; Baritaki, S.; Chatzaki, E. Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer. J. Clin. Med. 2021, 10, 2680. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Lambropoulou, M.; Balgkouranidou, I.; Nena, E.; Karaglani, M.; Nicolaidou, C.; Asimaki, A.; Konstantinidis, T.; Constantinidis, T.C.; Kolios, G.; et al. Gene promoter methylation and protein expression of BRMS1 in uterine cervix in relation to high-risk human papilloma virus infection and cancer. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317697557. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulou, M.; Fanidis, D.; Aidinis, V.; Chatzaki, E. ENPP2 Methylation in Health and Cancer. Int. J. Mol. Sci. 2021, 22, 11958. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, R37. [Google Scholar] [CrossRef] [Green Version]
- Brindley, D.N.; Tang, X.; Meng, G.; Benesch, M.G.K. Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5938. [Google Scholar] [CrossRef] [PubMed]
- Parris, T.Z.; Kovács, A.; Hajizadeh, S.; Nemes, S.; Semaan, M.; Levin, M.; Karlsson, P.; Helou, K. Frequent MYC coamplification and DNA hypomethylation of multiple genes on 8q in 8p11-p12-amplified breast carcinomas. Oncogenesis 2014, 3, e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Peng, Y.; Wang, J. Integrative analysis of DNA methylation and gene expression profiles identified potential breast cancer-specific diagnostic markers. Biosci. Rep. 2020, 40, BSR20201053. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Manolopoulos, V.G.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning. Cancers 2021, 13, 1677. [Google Scholar] [CrossRef] [PubMed]
- Ivan, J.; Patricia, G.; Agustriawan, D. In silico study of cancer stage-specific DNA methylation pattern in White breast cancer patients based on TCGA dataset. Comput. Biol. Chem. 2021, 92, 107498. [Google Scholar] [CrossRef]
- Popnikolov, N.K.; Dalwadi, B.H.; Thomas, J.D.; Johannes, G.J.; Imagawa, W.T. Association of autotaxin and lysophosphatidic acid receptor 3 with aggressiveness of human breast carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2012, 33, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef]
- Wang, S.-C.; Liao, L.-M.; Ansar, M.; Lin, S.-Y.; Hsu, W.-W.; Su, C.-M.; Chung, Y.-M.; Liu, C.-C.; Hung, C.-S.; Lin, R.-K. Automatic Detection of the Circulating Cell-Free Methylated DNA Pattern of GCM2, ITPRIPL1 and CCDC181 for Detection of Early Breast Cancer and Surgical Treatment Response. Cancers 2021, 13, 1375. [Google Scholar] [CrossRef]
- Wang, Z.; Jensen, M.A.; Zenklusen, J.C. A Practical Guide to The Cancer Genome Atlas (TCGA). Methods Mol. Biol. 2016, 1418, 111–141. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, J.; Bizet, M.; Desmedt, C.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Koch, A.; Larsimont, D.; Salgado, R.; Van den Eynden, G.; et al. DNA methylation-based immune response signature improves patient diagnosis in multiple cancers. J. Clin. Investig. 2017, 127, 3090–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.C.; Houseman, E.A.; King, J.E.; Christensen, B.C. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. BCR 2017, 19, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, J.I.J.; Knijnenburg, T.A.; Manughian-Peter, A.O.; Salomon, M.P.; Barkhoudarian, G.; Jalas, J.R.; Wilmott, J.S.; Hothi, P.; Wang, X.; Takasumi, Y.; et al. Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat. Commun. 2018, 9, 4627. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CG ID | Mβ Value Normal | Mβ Value BrCa | Δβ Value | Methylation in BrCa | Gene Region | FDR |
|---|---|---|---|---|---|---|
| Normal breast tissue vs. BrCa | ||||||
| cg00320790 | 0.96 | 0.95 | 0.01 | Down | Body | 5.97 × 10−4 |
| cg20048037 | 0.92 | 0.87 | 0.05 | Down | Body | 1.13 × 10−12 |
| cg09444531 | 0.77 | 0.79 | −0.02 | Up | Body | 5.16 × 10−3 |
| cg26078665 | 0.77 | 0.84 | −0.07 | Up | Body | 7.32 × 10−14 |
| cg23725583 | 0.85 | 0.92 | −0.06 | Up | Body | 1.03 × 10−15 |
| cg02534163 | 0.06 | 0.53 | −0.47 | Up | 1st Exon | 3.15 × 10−91 |
| cg04452959 | 0.03 | 0.44 | −0.41 | Up | TSS200 | 4.56 × 10−80 |
| cg02709432 | 0.09 | 0.57 | −0.48 | Up | TSS200 | 6.71 × 10−73 |
| cg02156680 | 0.04 | 0.44 | −0.39 | Up | TSS1500 | 9.18 × 10−72 |
| cg06998282 | 0.09 | 0.62 | −0.53 | Up | TSS1500 | 9.54 × 10−76 |
| Primary vs. Metastatic BrCa | ||||||
| cg20048037 | 0.87 | 0.82 | 0.06 | Down | Body | 3.99 × 10−2 |
| cg09444531 | 0.78 | 0.71 | 0.06 | Down | Body | 3.77 × 10−2 |
| cg26078665 | 0.86 | 0.79 | 0.07 | Down | Body | 9.09 × 10−4 |
| cg23725583 | 0.92 | 0.88 | 0.04 | Down | Body | 2.56 × 10−2 |
| cg02534163 | 0.55 | 0.74 | −0.19 | Up | 1st Exon | 1.26 × 10−4 |
| cg06998282 | 0.64 | 0.79 | −0.15 | Up | TSS1500 | 2.28 × 10−3 |
| Normal breast vs. stage I BrCa | ||||||
| cg20048037 | 0.92 | 0.89 | 0.03 | Down | Body | 4.35 × 10−4 |
| cg09444531 | 0.77 | 0.80 | −0.03 | Up | Body | 9.27 × 10−3 |
| cg26078665 | 0.78 | 0.86 | −0.08 | Up | Body | 1.07 × 10−7 |
| cg23725583 | 0.86 | 0.93 | −0.07 | Up | Body | 1.74 × 10−8 |
| cg02534163 | 0.06 | 0.55 | −0.48 | Up | 1st Exon | 2.45 × 10−49 |
| cg04452959 | 0.04 | 0.47 | −0.43 | Up | TSS200 | 5.53 × 10−38 |
| cg02709432 | 0.10 | 0.61 | −0.51 | Up | TSS200 | 1.14 × 10−38 |
| cg02156680 | 0.05 | 0.47 | −0.43 | Up | TSS1500 | 5.59 × 10−39 |
| cg06998282 | 0.10 | 0.66 | −0.56 | Up | TSS1500 | 2.93 × 10−35 |
| Early vs. Advanced BrCa | ||||||
| cg01243251 | 0.94 | 0.92 | 0.014 | Down | Body | 3.10 × 10−2 |
| cg20162626 | 0.75 | 0.69 | 0.051 | Down | Body | 3.12 × 10−3 |
| Compared Breast Groups | Fold Change | p-Value | FDR |
|---|---|---|---|
| Cancer_vs_Normal | −5.15 | 1.18 × 10−67 | 3.96 × 10−66 |
| StageI_vs_Normal | −5.46 | 6.28 × 10−54 | 3.43 × 10−52 |
| Advanced_vs_Early | 1.20 | 1.23 × 10−2 | 9.41 × 10−2 |
| BrCa vs. Normal | |||||
|---|---|---|---|---|---|
| Tissue | CG | Gene Region | Rho | FDR | Correlation |
| BrCa | cg02534163 | First Exon | −0.40 | 1.18 × 10−12 | Negative |
| cg06998282 | TSS1500 | −0.42 | 1.53 × 10−13 | Negative | |
| cg14409958 | TSS1500 | −0.42 | 2.10 × 10−13 | Negative | |
| Normal | cg09444531 | Body | 0.62 | 4.77 × 10−06 | Positive |
| cg23725583 | Body | −0.70 | 3.02 × 10−11 | Negative | |
| cg07236691 | Body | −0.55 | 1.27 × 10−08 | Negative | |
| cg14409958 | TSS1500 | −0.52 | 9.53 × 10−07 | Negative | |
| Stage I BrCa vs. Normal | |||||
| Stage I | cg02534163 | First Exon | −0.46 | 1.54 × 10−06 | Negative |
| cg04452959 | TSS200 | −0.44 | 3.74 × 10−06 | Negative | |
| cg14409958 | TSS1500 | −0.64 | 3.29 × 10−13 | Negative | |
| cg02156680 | TSS1500 | −0.42 | 1.28 × 10−05 | Negative | |
| cg06998282 | TSS1500 | −0.63 | 6.04 × 10−13 | Negative | |
| Normal | cg09444531 | Body | 0.64 | 3.25 × 10−08 | Positive |
| cg23725583 | Body | −0.66 | 1.36 × 10−08 | Negative | |
| cg07236691 | Body | −0.53 | 1.40 × 10−05 | Negative | |
| cg14409958 | TSS1500 | −0.51 | 4.25 × 10−05 | Negative | |
| Early vs. Advanced BrCa | |||||
| Early | cg02534163 | First Exon | −0.42 | 2.56 × 10−23 | Negative |
| cg06998282 | TSS1500 | −0.47 | 4.588 × 10−29 | Negative | |
| cg14409958 | TSS1500 | −0.46 | 5.37 × 10−28 | Negative | |
| Advanced | cg02534163 | First Exon | −0.43 | 1.23 × 10−09 | Negative |
| cg04452959 | TSS200 | −0.41 | 5.47 × 10−09 | Negative | |
| cg14409958 | TSS1500 | −0.48 | 4.72 × 10−12 | Negative | |
| cg06998282 | TSS1500 | −0.49 | 4.18 × 10−12 | Negative | |
| CG ID | Mβ Value Ductal Cancer | Mβ Value Lobular Cancer | Δβ Value | Methylation in Ductal Cancer | Gene Region | FDR |
|---|---|---|---|---|---|---|
| cg01243251 | 0.94 | 0.93 | 0.01 | Up | Body | 1.53 × 10−2 |
| cg20048037 | 0.86 | 0.90 | −0.04 | Down | Body | 1.11 × 10−2 |
| cg02156680 | 0.47 | 0.52 | −0.04 | Down | TSS1500 | 2.52 × 10−2 |
| CG ID | Mβ Value BrCa | Mβ Value Normal | ΔβValue | Methylation in BrCa | Location | FDR |
|---|---|---|---|---|---|---|
| cg07236691 | 0.583 | 0.817 | −0.234 | Down | Body | 3.04 × 10−3 |
| cg20048037 | 0.711 | 0.331 | 0.380 | Up | Body | 5.08 × 10−5 |
| cg20162626 | 0.489 | 0.814 | −0.325 | Down | Body | 3.18 × 10−4 |
| cg02534163 | 0.802 | 0.206 | 0.596 | Up | 1st Exon | 6.30 × 10−11 |
| cg04452959 | 0.620 | 0.020 | 0.599 | Up | TS200 | 2.51 × 10−13 |
| cg02156680 | 0.515 | 0.028 | 0.487 | Up | TSS1500 | 1.48 × 10−13 |
| cg06998282 | 0.772 | 0.057 | 0.715 | Up | TSS1500 | 1.61 × 10−8 |
| cg14409958 | 0.748 | 0.053 | 0.695 | Up | TSS1500 | 2.24 × 10−6 |
| Study Groups | Tissues | Age (Years) Median (Range) | Stage | Significance |
|---|---|---|---|---|
| 1. BrCa vs. Normal | 520 BrCa (primary and metastatic) | 49 (26–80) | 102 Stage I 264 Stage II 114 Stage III 40 Stage IV | Diagnosis |
| 185 Normal | 47 (26–80) | NR | ||
| 2. Primary vs. Metastatic BrCa | 132 PrimaryBrCa | 55 (47–55) | 22 Stage I 75 Stage II 35 Stage III | Diagnosis/Prognosis |
| 31 Metastatic BrCa | 54 (41–80) | 31 Stage IV | ||
| 3. Stage I BrCa vs. Normal | 136 Stage I BrCa | 54 (27–80) | 136 Stage I | Diagnosis/Prognosis |
| 111 Normal | 58 (29–80) | NR | ||
| 4. Early vs. Advanced BrCa | 521 EarlyBrCa | 58 (26–80) | 115 Stage I 406 Stage II | Diagnosis/Prognosis |
| 221 Advanced BrCa | 55 (27–80) | 221 Stage III |
| Group | Total | Adjuvant | Metastatic | Neoadjuvant | Control |
|---|---|---|---|---|---|
| N | 132 | 52 | 19 | 15 | 46 |
| Age | |||||
| Mean (±SD) | 57.7 (±13.9) | 58.7 (±12.0) | 61.9 (±9.8) | 55.5 (±16.6) | 55.6 (±13.7) |
| Median (range) | 59.0 (0.0–83.0) | 60.5 (27.0–80.0) | 65.0 (44.0–75.0) | 51.0 (29.0–79.0) | 57.0 (26.0–83.0) |
| Grade | |||||
| 1 | 10 | 10 | - | - | |
| 2 | 25 | 19 | - | 6 | |
| 3 | 30 | 16 | 8 | 6 | |
| N/A | 21 | 7 | 11 | 3 | |
| Stage | |||||
| I | 15 | 15 | - | - | |
| II | 28 | 28 | - | - | |
| III | 9 | 9 | - | - | |
| IV | 19 | - | 19 | - | |
| N/A | 15 | - | - | 15 | |
| Lymphnode status | |||||
| Negative | 27 | 24 | - | 3 | |
| Positive | 33 | 26 | - | 7 | |
| N/A | 26 | 2 | 19 | 5 | |
| Tumor size (before surgery) | |||||
| ≤2 | 30 | 25 | - | 5 | |
| >2 and ≤6.5 | 33 | 26 | - | 7 | |
| N/A | 23 | 1 | 19 | 3 | |
| Metastatic sites | |||||
| Lung | 12 | - | 12 | - | |
| Skin | 1 | - | 1 | - | |
| Distantlymphnodes | 5 | - | 5 | - | |
| Pancreas | 1 | - | 1 | - | |
| Bone | 9 | - | 9 | - | |
| Liver | 4 | - | 4 | - | |
| Pleural | 1 | - | 1 | - | |
| GENE | Primer Sequence (5′–3′) | Annealing Temperature (°C) | Product Length | Genomic Loci |
|---|---|---|---|---|
| ENPP2 | MET F: CGTTTTTTTATTTGATACGATTGGAACGA MET R: CAAAACCTCAAAACAATACACTCCGTAA | 60 | 117bp | Chr8: 120650976- 120651092 (+1 strand) |
| ACTB | F: TGGTGATGGAGGAGGTTTAGTAAG R: AACCAATAAAACCTACTCCTCCC | 60 | 134bp | chr7: 5558705– 5558838 (−1 strand) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panagopoulou, M.; Drosouni, A.; Fanidis, D.; Karaglani, M.; Balgkouranidou, I.; Xenidis, N.; Aidinis, V.; Chatzaki, E. ENPP2 Promoter Methylation Correlates with Decreased Gene Expression in Breast Cancer: Implementation as a Liquid Biopsy Biomarker. Int. J. Mol. Sci. 2022, 23, 3717. https://doi.org/10.3390/ijms23073717
Panagopoulou M, Drosouni A, Fanidis D, Karaglani M, Balgkouranidou I, Xenidis N, Aidinis V, Chatzaki E. ENPP2 Promoter Methylation Correlates with Decreased Gene Expression in Breast Cancer: Implementation as a Liquid Biopsy Biomarker. International Journal of Molecular Sciences. 2022; 23(7):3717. https://doi.org/10.3390/ijms23073717
Chicago/Turabian StylePanagopoulou, Maria, Andrianna Drosouni, Dionysiοs Fanidis, Makrina Karaglani, Ioanna Balgkouranidou, Nikolaos Xenidis, Vassilis Aidinis, and Ekaterini Chatzaki. 2022. "ENPP2 Promoter Methylation Correlates with Decreased Gene Expression in Breast Cancer: Implementation as a Liquid Biopsy Biomarker" International Journal of Molecular Sciences 23, no. 7: 3717. https://doi.org/10.3390/ijms23073717
APA StylePanagopoulou, M., Drosouni, A., Fanidis, D., Karaglani, M., Balgkouranidou, I., Xenidis, N., Aidinis, V., & Chatzaki, E. (2022). ENPP2 Promoter Methylation Correlates with Decreased Gene Expression in Breast Cancer: Implementation as a Liquid Biopsy Biomarker. International Journal of Molecular Sciences, 23(7), 3717. https://doi.org/10.3390/ijms23073717