
Midecamycin Is Inactivated by Several Different Sugar Moieties at Its Inactivation Site


Abstract
1. Introduction
2. Results
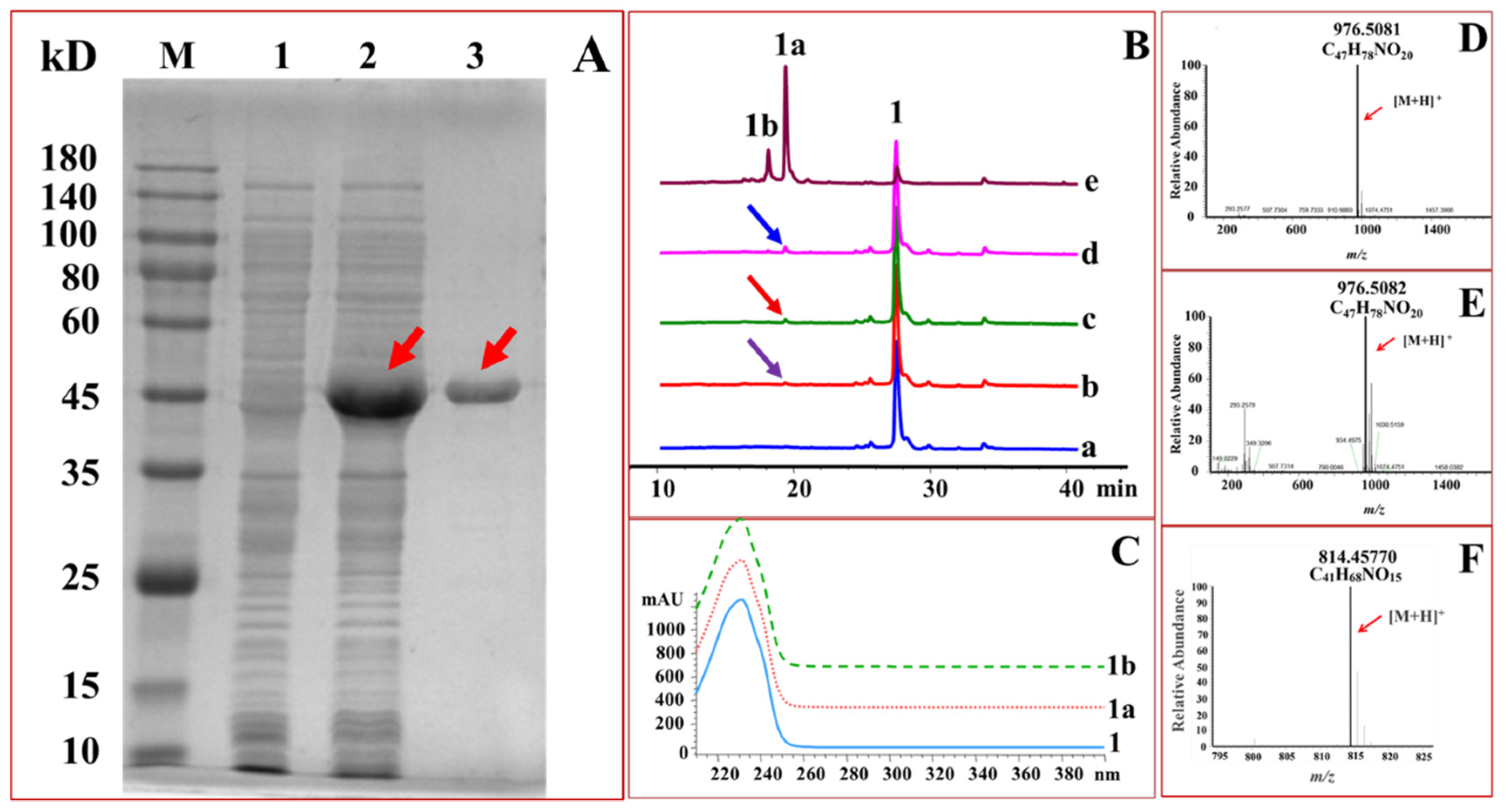
2.1. Expression and Purification of GTs
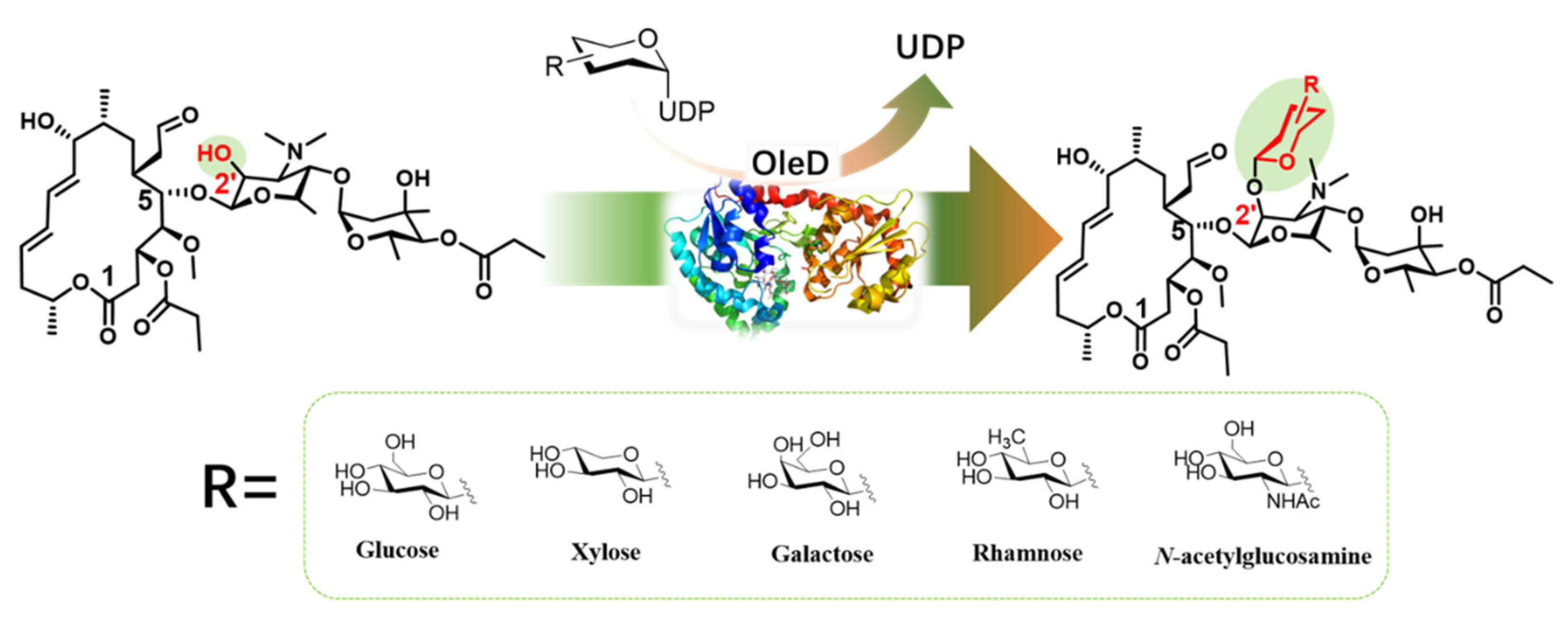
2.2. Glucosylation of Midecamycin
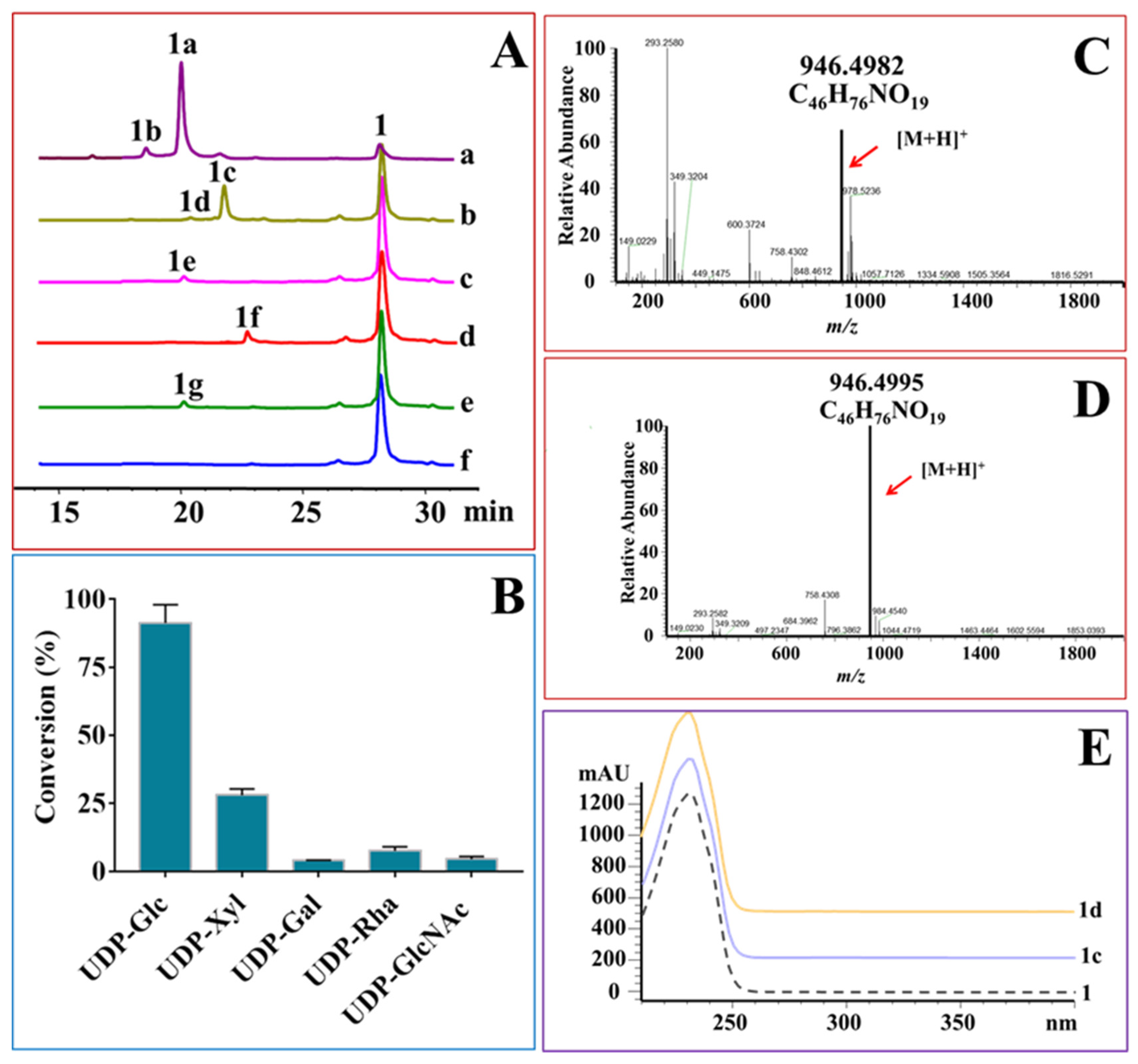
2.3. OleD-Mediated Glycodiversificaion of Midecamycin
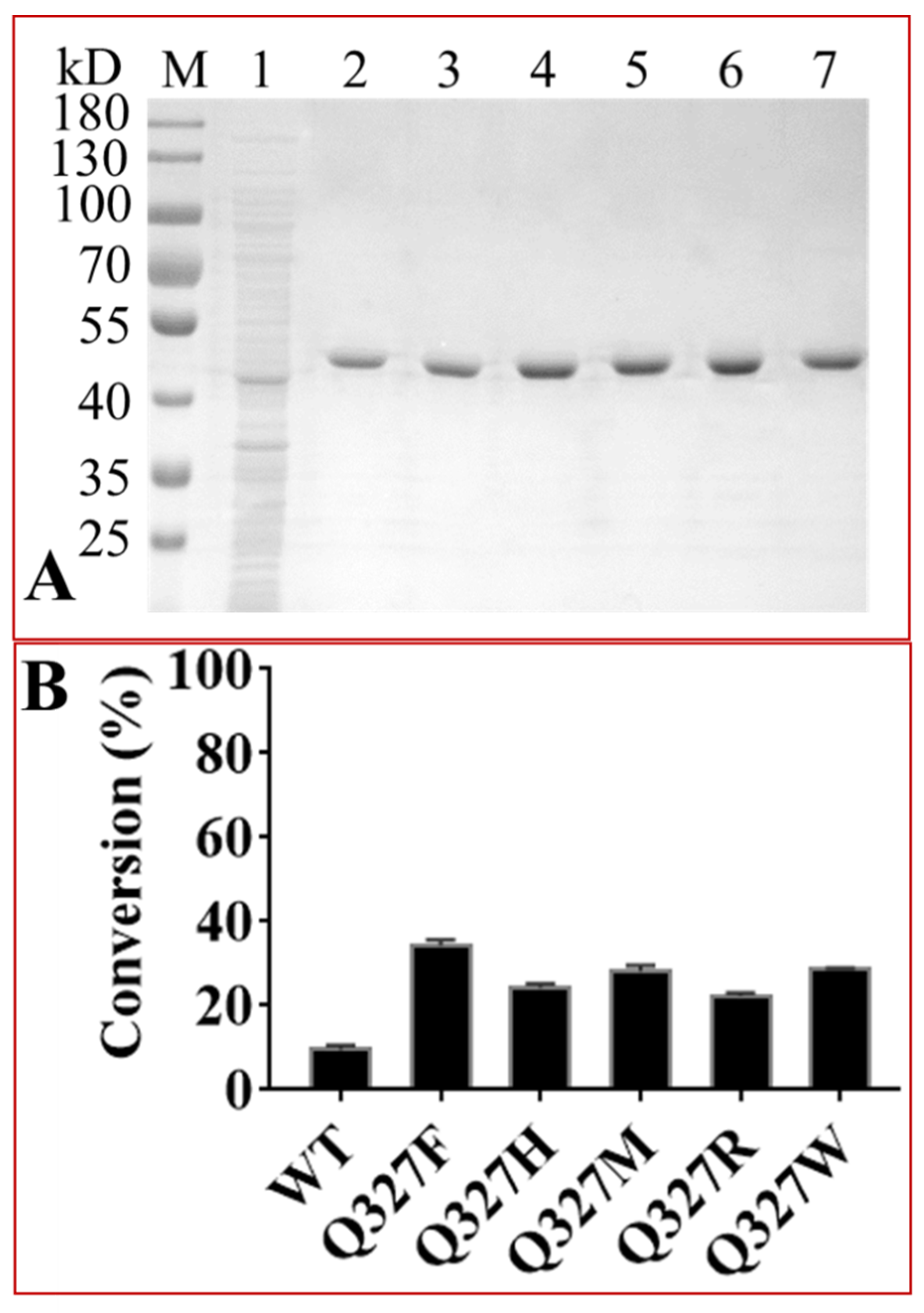
2.4. Protein Engineering of OleD to Enhance Its Catalytic Efficiencies
2.5. Water Solubility of Midecamycin 2′-O-Glucopyranoside
2.6. Antimicrobial Activities of Midecamycin 2′-O-Glycosides
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Plasmids and Strains
4.3. Protein Expression and Purification
4.4. Glycosylation Assays
4.5. Condition Optimization for OleD-Catalyzed Reactions
4.6. Molecular Docking of Ligands with Proteins
4.7. Directed Mutations of OleD
4.8. Water Solubility Assay
4.9. Evaluation of Antimicrobial Activity of Midecamycin 2′-O-Glycosides
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arenz, S.; Ramu, H.; Gupta, P.; Berninghausen, O.; Beckmann, R.; Vazquez-Laslop, N.; Mankin, A.S.; Wilson, D.N. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat. Commun. 2014, 5, 3501. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Hwang, I.Y.; Jo, Y.J.; Choi, S.H.; Lee, E.Y. Enzymatic synthesis of amentoflavone glycoside using recombinant oleandomycin glycosyltransferase. J. Ind. Eng. Chem. 2015, 25, 304–307. [Google Scholar] [CrossRef]
- Choi, S.H.; Ryu, M.; Yoon, Y.J.; Kim, D.M.; Lee, E.Y. Glycosylation of various flavonoids by recombinant oleandomycin glycosyltransferase from Streptomyces antibioticus in batch and repeated batch modes. Biotechnol. Lett. 2012, 34, 499–505. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, B.; Ren, X.; Wang, J. Universal response method for the quantitative analysis of multi-components in josamycin and midecamycin using liquid chromatography coupled with charged aerosol detector. J. Pharm. Biomed. Anal. 2021, 192, 113679. [Google Scholar] [CrossRef]
- Zhao, Z.; Jin, L.; Xu, Y.; Zhu, D.; Liu, Y.; Liu, C.; Lei, P. Synthesis and antibacterial activity of a series of novel 9-O-acetyl-4′-substituted 16-membered macrolides derived from josamycin. Bioorg. Med. Chem. Lett. 2014, 24, 480–484. [Google Scholar] [CrossRef]
- Wang, N.; Zhou, Y.; Zhang, H.; Liu, Y. In vitro activities of acetylmidecamycin and other antimicrobials against human macrolide-resistant Mycoplasma pneumoniae isolates. J. Antimicrob. Chemother. 2020, 75, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wang, Y.; Liu, J.; He, W. The regulatory genes involved in spiramycin and bitespiramycin biosynthesis. Microbiol. Res. 2020, 240, 126532. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, X.; Dai, J.; Wang, Y.; He, W. Engineering of leucine-responsive regulatory protein improves spiramycin and bitespiramycin biosynthesis. Microb. Cell Fact. 2019, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, Y.; Li, W.C.; Tang, B.X.; Li, J.; Zang, Y. Roxithromycin attenuates bleomycin-induced pulmonary fibrosis by targeting senescent cells. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Capecchi, M.; Lazzerini, P.E. Roxithromycin-associated acute thrombocytopenia. Am. J. Case Rep. 2021, 22, e932039. [Google Scholar] [CrossRef]
- Sidhu, H.; Bae, H.S.; Ogram, A.; O’Connor, G.; Yu, F. Azithromycin and ciprofloxacin can promote antibiotic resistance in biosolids and biosolids-amended soils. Appl. Environ. Microbiol. 2021, 87, e0037321. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, R.; Yu, P.; Li, Y.; Pan, Q.; Li, Y. The macrolide antibiotic azithromycin potently inhibits hepatitis E virus in cell culture models. Int. J. Antimicrob. Agents 2021, 58, 106383. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nam, S.; Kim, E.; Jeon, H.; Lee, K.; Xie, K. Identification of erythromycin and clarithromycin metabolites formed in chicken liver microsomes using liquid chromatography-high-resolution mass spectrometry. Foods 2021, 10, 1504. [Google Scholar] [CrossRef] [PubMed]
- Saruuljavkhlan, B.; Yamaoka, Y. Benefits of a molecular-based method for the detection of clarithromycin-resistant Helicobacter pylori. Gut Liver 2021, 15, 487–489. [Google Scholar] [CrossRef] [PubMed]
- Breiner-Goldstein, E.; Eyal, Z.; Matzov, D.; Halfon, Y.; Cimicata, G.; Baum, M.; Rokney, A.; Ezernitchi, A.V.; Lowell, A.N.; Schmidt, J.J.; et al. Ribosome-binding and anti-microbial studies of the mycinamicins, 16-membered macrolide antibiotics from Micromonospora griseorubida. Nucleic Acids Res. 2021, 49, 9560–9573. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Laslop, N.; Mankin, A.S. How macrolide antibiotics work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and outlook on enzyme-mediated macrolide resistance. Front. Microbiol. 2018, 9, 1942. [Google Scholar] [CrossRef]
- Matsuoka, M.; Sasaki, T. Inactivation of macrolides by producers and pathogens. Curr. Drug Targets Infect. Disord. 2004, 4, 217–240. [Google Scholar] [CrossRef]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Peterson, E.; Kaur, P. Antibiotic resistance mechanisms in bacteria: Relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef]
- Egorov, A.M.; Ulyashova, M.M.; Rubtsova, M.Y. Bacterial enzymes and antibiotic resistance. Acta Nat. 2018, 10, 33–48. [Google Scholar] [CrossRef]
- Hopwood, D.A. How do antibiotic-producing bacteria ensure their self-resistance before antibiotic biosynthesis incapacitates them? Mol. Microbiol. 2007, 63, 937–940. [Google Scholar] [CrossRef]
- Kuo, M.-S.; Chirby, D.; Argoudelis, A.; Cialdella, J.; Coats, J.; Marshall, V. Microbial glycosylation of erythromycin A. Antimicrob. Agents Chemother. 1989, 33, 2089–2091. [Google Scholar] [CrossRef]
- Jenkins, G.; Cundliffe, E. Cloning and characterization of two genes from Streptomyces lividans that confer inducible resistance to lincomycin and macrolide antibiotics. Gene 1991, 108, 55–62. [Google Scholar] [CrossRef]
- Cundliffe, E. Resistance to macrolides and lincosamides in Streptomyces lividans and to aminoglycosides in Micromonospora purpurea. Gene 1992, 115, 75–84. [Google Scholar] [CrossRef]
- Cundliffe, E. Glycosylation of macrolide antibiotics in extracts of Streptomyces lividans. Antimicrob. Agents Chemother. 1992, 36, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Vilches, C.; Hernandez, C.; Mendez, C.; Salas, J.A. Role of glycosylation and deglycosylation in biosynthesis of and resistance to oleandomycin in the producer organism, Streptomyces antibioticus. J. Bacteriol. 1992, 174, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Olano, C.; Méndez, C.; Salas, J. Characterization of a Streptomyces antibioticus gene cluster encoding a glycosyltransferase involved in oleandomycin inactivation. Gene 1993, 134, 139–140. [Google Scholar] [CrossRef]
- Quirós, L.M.; Salas, J.A. Biosynthesis of the macrolide oleandomycin by Streptomyces antibioticus: Purification and kinetic characterization of an oleandomycin glucosyltransferase. J. Biol. Chem. 1995, 270, 18234–18239. [Google Scholar] [CrossRef]
- Quirós, L.M.; Aguirrezabalaga, I.; Olano, C.; Méndez, C.; Salas, J.A. Two glycosyltransferases and a glycosidase are involved in oleandomycin modification during its biosynthesis by Streptomyces antibioticus. Mol. Microbiol. 1998, 28, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Quirós, L.M.; Carbajo, R.J.; Salas, J.A. Inversion of the anomeric configuration of the transferred sugar during inactivation of the macrolide antibiotic oleandomycin catalyzed by a macrolide glycosyltransferase. FEBS Lett. 2000, 476, 186–189. [Google Scholar] [CrossRef]
- Schulman, M.; Doherty, P.; Arison, B. Microbial conversion of avermectins by Saccharopolyspora erythraea: Glycosylation at C-4′ and C-4″. Antimicrob. Agents Chemother. 1993, 37, 1737–1741. [Google Scholar] [CrossRef][Green Version]
- Yazawa, K.; Mikami, Y.; Sakamoto, T.; Ueno, Y.; Morisaki, N.; Iwasaki, S.; Furihata, K. Inactivation of the macrolide antibiotics erythromycin, midecamycin, and rokitamycin by pathogenic Nocardia species. Antimicrob. Agents Chemother. 1994, 38, 2197–2199. [Google Scholar] [CrossRef][Green Version]
- Morisaki, N.; Hashimoto, Y.; Furihata, K.; Yazawa, K.; Tamura, M.; Mikami, Y. Glycosylative inactivation of chalcomycin and tylosin by a clinically isolated Nocardia asteroides strain. J. Antibiot. 2001, 54, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, J.; Mizoue, K.; Morimoto, S.; Omura, S. Microbial glycosylation of macrolide antibiotics by Streptomyces hygroscopicus ATCC 31080 and distribution of a macrolide glycosyl transferase in several Streptomyces strains. J. Antibiot. 1996, 49, 1110–1118. [Google Scholar] [CrossRef][Green Version]
- Gourmelen, A.; Blondelet-Rouault, M.H.; Pernodet, J.L. Characterization of a glycosyl transferase inactivating macrolides, encoded by gimA from Streptomyces ambofaciens. Antimicrob. Agents Chemother. 1998, 42, 2612–2619. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, L.; Yue, X.-J.; Tang, Y.-J.; Wu, C.; Li, Y.-Z. Effects of glycosylation on the bioactivity of rapamycin. Appl. Microbiol. Biotechnol. 2020, 104, 9125–9134. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Z.; Li, Z.-F.; Chen, Q.; Li, Y.-Y.; Gong, Y.; Yue, X.-J.; Sheng, D.-H.; Zhang, Y.-M.; Wu, C.; et al. Phylogeny-guided characterization of glycosyltransferases for epothilone glycosylation. Microb. Biotechnol. 2019, 12, 763–774. [Google Scholar] [CrossRef]
- Yang, M.; Proctor, M.R.; Bolam, D.N.; Errey, J.C.; Field, R.A.; Gilbert, H.J.; Davis, B.G. Probing the breadth of macrolide glycosyltransferases: in vitro remodeling of a polyketide antibiotic creates active bacterial uptake and enhances potency. J. Am. Chem. Soc. 2005, 127, 9336–9337. [Google Scholar] [CrossRef] [PubMed]
- Bolam, D.N.; Roberts, S.; Proctor, M.R.; Turkenburg, J.P.; Dodson, E.J.; Martinez-Fleites, C.; Yang, M.; Davis, B.G.; Davies, G.J.; Gilbert, H.J. The crystal structure of two macrolide glycosyltransferases provides a blueprint for host cell antibiotic immunity. Proc. Natl. Acad. Sci. USA 2007, 104, 5336–5341. [Google Scholar] [CrossRef] [PubMed]
- Quiros, L.M.; Carbajo, R.J.; Brana, A.F.; Salas, J.A. Glycosylation of macrolide antibiotics. Purification and kinetic studies of a macrolide glycosyltransferase from Streptomyces antibioticus. J. Biol. Chem. 2000, 275, 11713–11720. [Google Scholar] [PubMed]
- Quiros, L.M.; Hernandez, C.; Salas, J.A. Purification and characterization of an extracellular enzyme from Streptomyces antibioticus that converts inactive glycosylated oleandomycin into the active antibiotic. Eur. J. Biochem. 1994, 222, 129–135. [Google Scholar] [CrossRef]
- Arsic, B.; Barber, J.; Čikoš, A.; Mladenovic, M.; Stankovic, N.; Novak, P. 16-membered macrolide antibiotics: A review. Int. J. Antimicrob. Agents 2018, 51, 283–298. [Google Scholar] [CrossRef]
- Cong, L.; Piepersberg, W. Cloning and characterization of genes encoded in dTDP-D-mycaminose biosynthetic pathway from a midecamycin-producing strain, Streptomyces mycarofaciens. Acta Biochim. Biophys. Sin. 2007, 39, 187–193. [Google Scholar] [CrossRef]
- Cui, W.; Ma, S. Recent advances in the field of 16-membered macrolide antibiotics. Mini-Rev. Med. Chem. 2011, 11, 1009–1018. [Google Scholar] [CrossRef]
- Schlegel, L.; Merad, B.; Rostane, H.; Broc, V.; Bouvet, A. In vitro activity of midecamycin diacetate, a 16-membered macrolide, against Streptococcus pyogenes isolated in France, 1995-1999. Clin. Microbiol. Infect. 2001, 7, 362–366. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, J.; Duan, X.; Lu, L.; Zhang, K.; Gao, Y.; Dong, L.; Sun, H. Facile fabrication of three-dimensional graphene microspheres using β-cyclodextrin aggregates as substrates and their application for midecamycin sensing. RSC Adv. 2015, 5, 77469–77477. [Google Scholar] [CrossRef]
- Song, X.; Zhou, T.; Li, J.; Zhang, M.; Xie, J.; He, L. Determination of ten macrolide drugs in environmental water using molecularly imprinted solid-phase extraction coupled with liquid chromatography-tandem mass spectrometry. Molecules. 2018, 23, 1172. [Google Scholar] [CrossRef]
- Wang, Z.; Song, X.; Zhou, T.; Bian, K.; Zhang, F.; He, L.; Liu, Q. Simultaneous determination of ten macrolides drugs in feeds by high performance liquid chromatography with evaporation light scattering detection. RSC Adv. 2015, 5, 1491–1499. [Google Scholar] [CrossRef]
- Tang, L.; McDaniel, R. Construction of desosamine containing polyketide libraries using a glycosyltransferase with broad substrate specificity. Chem. Biol. 2001, 8, 547–555. [Google Scholar] [CrossRef][Green Version]
- Isiorho, E.A.; Jeon, B.-S.; Kim, N.H.; Liu, H.-W.; Keatinge-Clay, A.T. Structural studies of the spinosyn forosaminyltransferase, SpnP. Biochemistry 2014, 53, 4292–4301. [Google Scholar] [CrossRef]
- Gaisser, S.; Carletti, I.; Schell, U.; Graupner, P.R.; Sparks, T.C.; Martin, C.J.; Wilkinson, B. Glycosylation engineering of spinosyn analogues containing an L-olivose moiety. Org. Biomol. Chem. 2009, 7, 1705–1708. [Google Scholar] [CrossRef]
- Nguyen, H.C.; Karray, F.; Lautru, S.; Gagnat, J.; Lebrihi, A.; Huynh, T.D.; Pernodet, J.L. Glycosylation steps during spiramycin biosynthesis in Streptomyces ambofaciens: Involvement of three glycosyltransferases and their interplay with two auxiliary proteins. Antimicrob. Agents Chemother. 2010, 54, 2830–2839. [Google Scholar] [CrossRef]
- Skinner, M.; Taylor, R.B.; Kanfer, I. The pH-stability and acid degradation of the macrolide antibiotic, josamycin. Eur. J. Pharm. Sci. 1993, 1, 61–72. [Google Scholar] [CrossRef]
- Omoto, S.; Ogino, H.; Iwamatsu, K.; Inouye, S. Modification of the macrolide antibiotic midecamycin. III. Formation of neoisomidecamycin. J. Antibiot. 1982, 35, 1521–1526. [Google Scholar] [CrossRef]
- Gantt, R.W.; Peltier-Pain, P.; Singh, S.; Zhou, M.; Thorson, J.S. Broadening the scope of glycosyltransferase-catalyzed sugar nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 7648–7653. [Google Scholar] [CrossRef]
- Chang, A.; Singh, S.; Phillips, G.N., Jr.; Thorson, J.S. Glycosyltransferase structural biology and its role in the design of catalysts for glycosylation. Curr. Opin. Biotechnol. 2011, 22, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P. The macrolide antibiotic renaissance. Brit. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef] [PubMed]
- Malmierca, M.G.; Pérez-Victoria, I.; Martín, J.; Reyes, F.; Méndez, C.; Salas, J.A.; Olano, C. New sipanmycin analogues generated by combinatorial biosynthesis and mutasynthesis approaches relying on the substrate flexibility of key enzymes in the biosynthetic pathway. Appl. Environ. Microbiol. 2020, 86, e02453-19. [Google Scholar] [CrossRef] [PubMed]
- Hara, O.; Hutchinson, C.R. A macrolide 3-O-acyltransferase gene from the midecamycin-producing species Streptomyces mycarofaciens. J. Bacteriol. 1992, 174, 5141–5144. [Google Scholar] [CrossRef]
- Xue, Y.; Zhao, L.; Liu, H.-W.; Sherman, D.H. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity. Proc. Natl. Acad. Sci. USA 1998, 95, 12111–12116. [Google Scholar] [CrossRef] [PubMed]
- Waldron, C.; Matsushima, P.; Rosteck, P.R.; Broughton, M.C.; Turner, J.; Madduri, K.; Crawford, K.P.; Merlo, D.J.; Baltz, R.H. Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa. Chem. Biol. 2001, 8, 487–499. [Google Scholar] [CrossRef]
- Wang, X.-N.; Hong, L.-L.; Kong, J.-Q. Diacerein as a promising acyl donor in biosynthetic acetyl-CoA and glycosyl esters mediated by a multifunctional maltose O-acetyltransferase from Escherichia coli. J. Agric. Food Chem. 2021, 69, 6623–6635. [Google Scholar] [CrossRef]
- Veseli, A.; Žakelj, S.; Kristl, A. A review of methods for solubility determination in biopharmaceutical drug characterization. Drug Dev. Ind. Pharm. 2019, 45, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Yang, C.; Zhao, X.; Wang, Y. Antimicrobial activity of bitespiramycin, a new genetically engineered macrolide. Bioorg. Med. Chem. Lett. 2017, 27, 4576–4577. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a | 1c | |||
|---|---|---|---|---|
| Position | δC | δH (mult, J Hz) | δC | δH (mult, J Hz) |
| 1 | 172.3 | 172.2 | ||
| 2 | 37.9 | 2.65 (dd, 12.3, 12.0) | 37.9 | 2.65 (overlap) |
| 2.41 (dd, 13.2, 5.4) | 2.37 (dd, 12.3, 7.6) | |||
| 3 | 71.1 | 5.09–5.14 (m) | 71.1 | 5.08–5.13 (m) |
| 4 | 85.7 | 3.54 (dd, 10.2, 6.0) | 85.9 | 3.38 (overlap) |
| 5 | 76.8 | 3.87 (overlap) | 76.3 | 3.89 (overlap) |
| 6 | 30.2 | 2.13–2.21 (m) | 30.1 | 2.14–2.23 (m) |
| 7 | 31.6 | 1.48–1.56 (m) | 32.6 | 1.52–1.61 (m) |
| 1.07–1.13 (m) | 0.98–1.03 (m) | |||
| 8 | 35.2 | 1.86–1.95 (m) | 35.1 | 1.92–2.00 (m) |
| 9 | 74.1 | 4.23 (dd, 9.6, 5.4) | 74.1 | 4.22 (dd, 9.6, 7.6) |
| 10 | 128.8 | 5.64 (dd, 15.6, 9.6) | 128.6 | 5.65 (dd, 15.3, 5.8) |
| 11 | 136.6 | 6.58 (dd, 16.0, 10.2) | 136.7 | 6.59 (dd, 15.2, 10.4) |
| 12 | 133.9 | 6.10 (dd,15.0, 10.8) | 133.8 | 6.11 (dd, 14.9, 10.6) |
| 13 | 133.2 | 5.71–5.78 (m) | 133.3 | 5.71–5.79 (m) |
| 14 | 41.8 | 2.13–2.21 (m) | 42.8 | 2.47–2.51 (m) |
| 2.45–2.50 (m) | 2.16–2.22 (m) | |||
| 15 | 70.6 | 4.94 (dd, 13.8, 6.6) | 70.7 | 4.93 (dd, 10.0, 6.5) |
| 16 | 20.6 | 1.25 (d, 6.0) | 20.9 | 1.27 (d, 5.6) |
| 17 | 43.5 | 2.82–2.90 (m) | 43.4 | 2.83 (dd, 18.6, 11.3) |
| 2.36 (dd, 18.0, 7.8) | 2.36 (overlap) | |||
| 18 | 203.5 | 9.66 (s) | 203.5 | 9.66 (s) |
| 19 | 15.3 | 0.99 (t, 5.4) | 15.3 | 0.99 (d, 5.2) |
| 20 | 175.8 | 175.8 | ||
| 21 | 28.5 | 2.43–2.49 (m) | 28.5 | 2.41–2.49 (m) |
| 22 | 9.6 | 1.19 (overlap) | 9.6 | 1.19 (overlap) |
| 23 | 62.9 | 3.62 (s) | 62.5 | 3.58 (s) |
| 1′ | 102.7 | 4.69 | 102.1 | 4.69 (d, 7.1) |
| 2′ | 82.3 | 3.42 (dd, 7.8, 6.0) | 81.5 | 3.48 (dd, 8.9, 5.6) |
| 3′ | 69.7 | 2.87 (overlap) | 69.9 | 2.98 (overlap) |
| 4′ | 80.4 | 3.63 (overlap) | 78.5 | 3.70 (dd, 9.6, 7.3) |
| 5′ | 73.5 | 3.54–3.60 (m) | 73.5 | 3.52–3.56 (m) |
| 6′ | 19.3 | 1.34 (d, 6.0) | 19.3 | 1.36 (d, 6.2) |
| 7′ and 8′ | 41.8 | 2.59(s) | 41.9 | 2.68 (s) |
| 1″ | 99.0 | 5.16 (overlap) | 98.7 | 5.17 (overlap) |
| 2″ | 43.0 | 2.06 (d, 15.0) | 42.8 | 2.06 (dd, 14.5, 4.6) |
| 1.96 (dd, 14.4, 4.8) | 1.97 (d, 14.1) | |||
| 3″ | 70.4 | 70.6 | ||
| 4″ | 78.6 | 4.60 (d, 10.2) | 78.5 | 4.62 (d, 9.8) |
| 5″ | 65.0 | 4.43–4.47 (m) | 65.1 | 4.37 (overlap) |
| 6″ | 17.9 | 1.11 (s) | 17.9 | 1.13 (d, 2.3) |
| 7″ | 26.8 | 1.13 (s) | 26.5 | 1.14 (s) |
| 8″ | 175.8 | 175.8 | ||
| 9″ | 28.3 | 2.45 (overlap) | 28.3 | 2.46 (overlap) |
| 10″ | 9.6 | 1.20 | 9.6 | 1.21 (overlap) |
| 1‴ | 107.8 | 4.40 (dd, 7.8, 7.2) | 107.8 | 4.88 (d, 7.8) |
| 2‴ | 76.5 | 3.21 (dd, 8.4, 8.4) | 76.1 | 3.21 (dd, 9.0, 7.6) |
| 3‴ | 77.8 | 3.38 (dd, 9.0, 9.0) | 78.0 | 3.35 (overlap) |
| 4‴ | 71.3 | 3.34 (overlap) | 71.1 | 3.46 (overlap) |
| 5‴ | 78.5 | 3.29 (overlap) | 67.5 | 3.90 (overlap) |
| 3.2 (overlap) | ||||
| 6‴ | 62.9 | 3.89 (overlap) | ||
| 3.76 (dd, 12.0, 4.8) | ||||
| Substrate | Km (mM) | Vmax (mM/min) |
|---|---|---|
| UDP-Glc | 1.933 ± 0.124 | 0.080 ± 0.002 |
| midecamycin | 0.626 ± 0.106 | 11.120 ± 0.830 |
| Strain | MIC (μg/mL) | |||
|---|---|---|---|---|
| Midecamycin | 1a | 1c | 1g | |
| Bacillus intestinalis strain T30 | 0.5 | — | — | — |
| Bacillus subtilis strain 168 | 1 | — | — | — |
| Staphylococcus aureus | 1 | — | — | — |
| Streptococcus pneumoniae | 0.25 | — | — | — |
| Pseudomonas aeruginosa PAO1 | — | — | — | — |
| Escherichia coli DH5α | — | — | — | — |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, R.; Hong, L.-L.; Jiang, Z.-K.; Li, K.-M.; He, W.-Q.; Kong, J.-Q. Midecamycin Is Inactivated by Several Different Sugar Moieties at Its Inactivation Site. Int. J. Mol. Sci. 2021, 22, 12636. https://doi.org/10.3390/ijms222312636
Lin R, Hong L-L, Jiang Z-K, Li K-M, He W-Q, Kong J-Q. Midecamycin Is Inactivated by Several Different Sugar Moieties at Its Inactivation Site. International Journal of Molecular Sciences. 2021; 22(23):12636. https://doi.org/10.3390/ijms222312636
Chicago/Turabian StyleLin, Ru, Li-Li Hong, Zhong-Ke Jiang, Ke-Meng Li, Wei-Qing He, and Jian-Qiang Kong. 2021. "Midecamycin Is Inactivated by Several Different Sugar Moieties at Its Inactivation Site" International Journal of Molecular Sciences 22, no. 23: 12636. https://doi.org/10.3390/ijms222312636
APA StyleLin, R., Hong, L.-L., Jiang, Z.-K., Li, K.-M., He, W.-Q., & Kong, J.-Q. (2021). Midecamycin Is Inactivated by Several Different Sugar Moieties at Its Inactivation Site. International Journal of Molecular Sciences, 22(23), 12636. https://doi.org/10.3390/ijms222312636