
Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
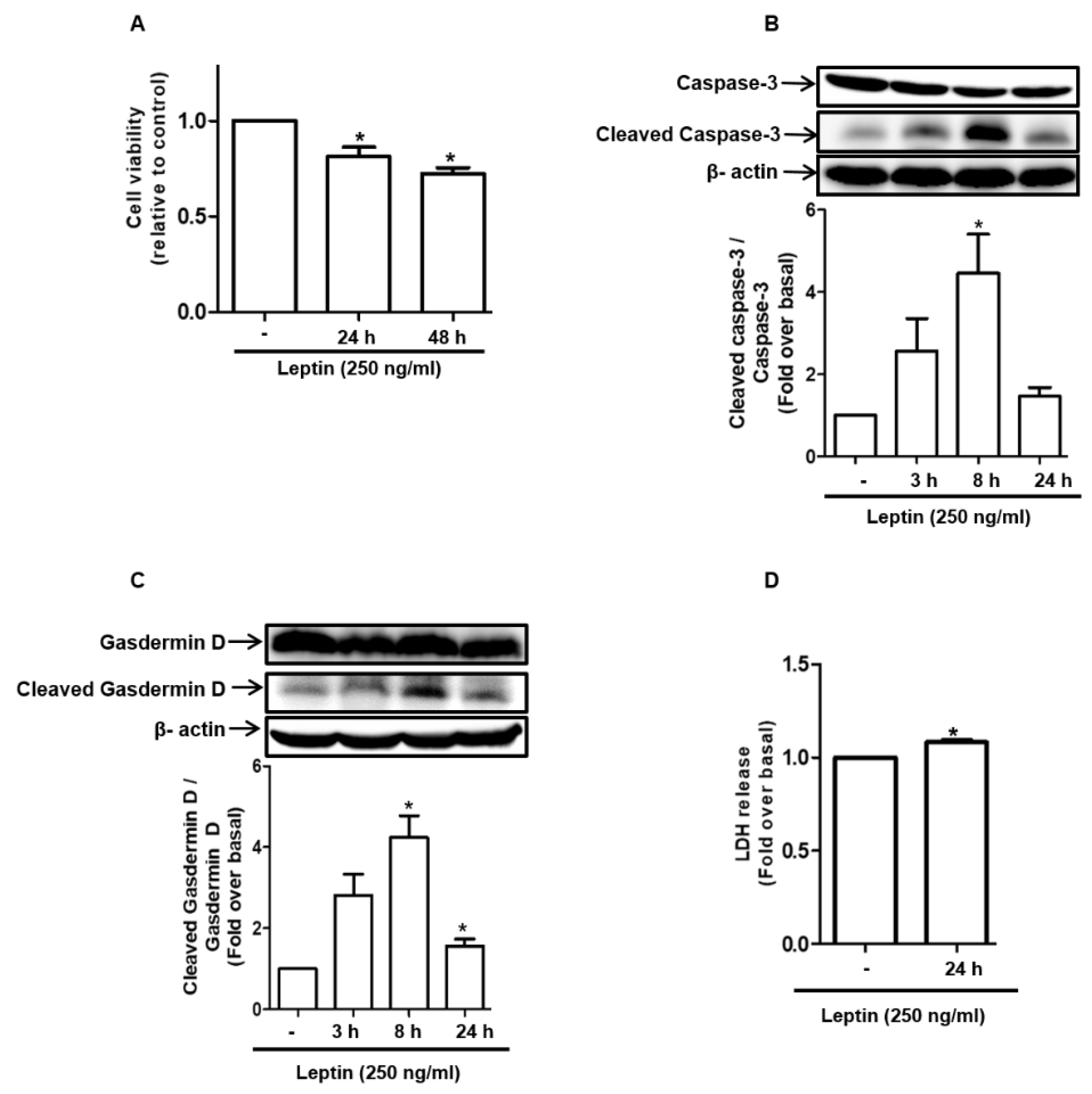
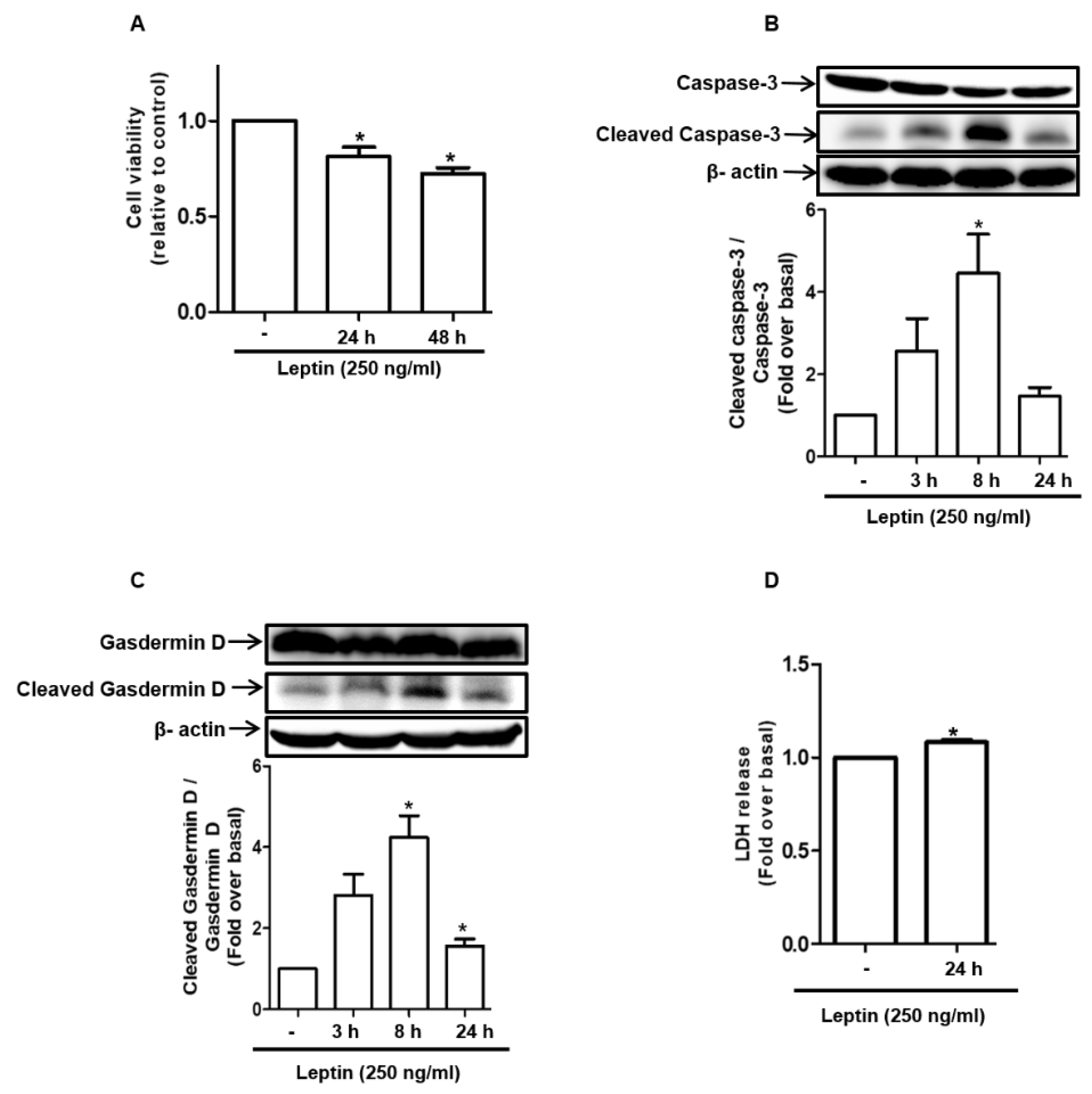
2.1. Leptin Induces Cell Death in Rat Hepatocytes
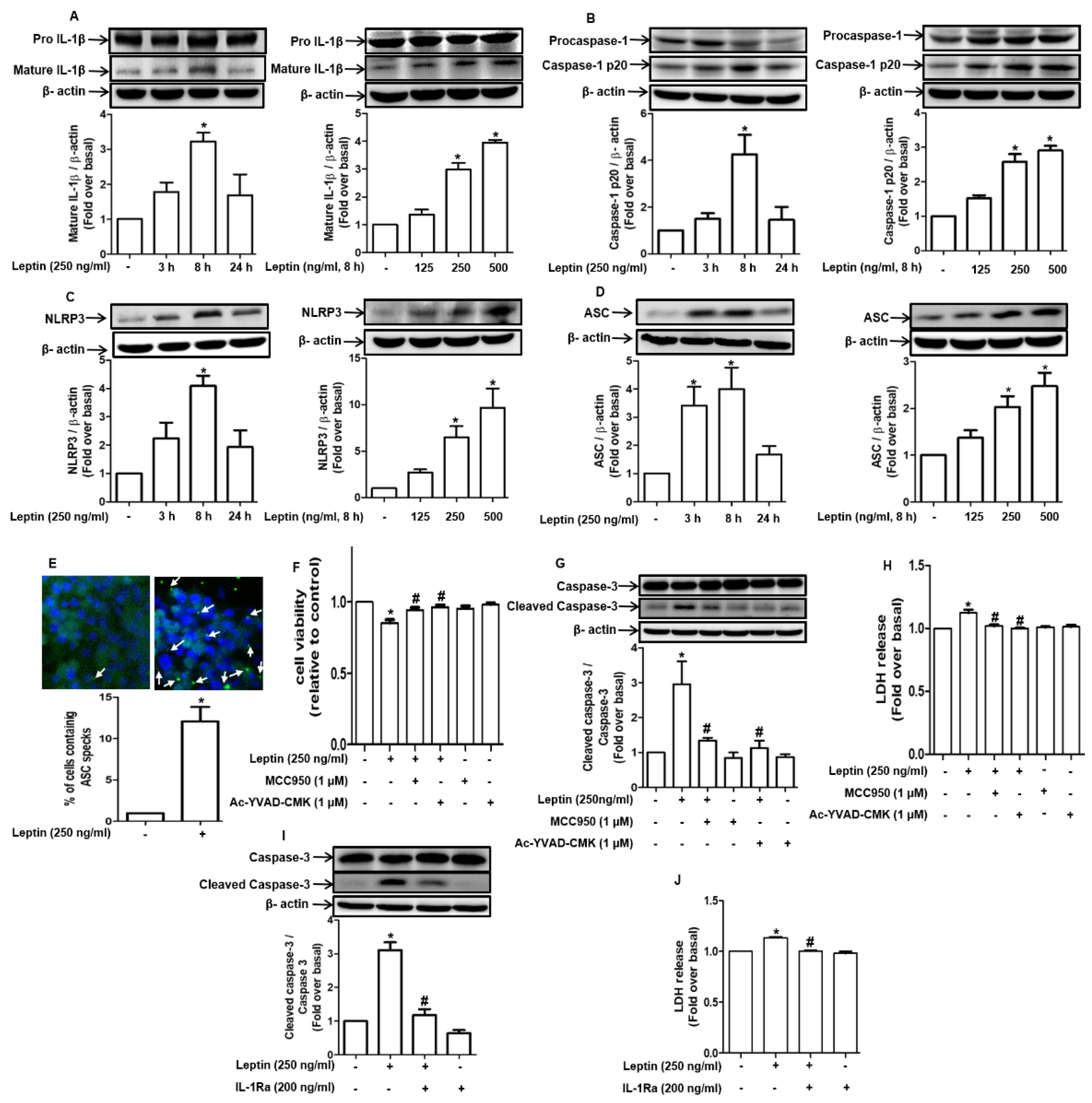
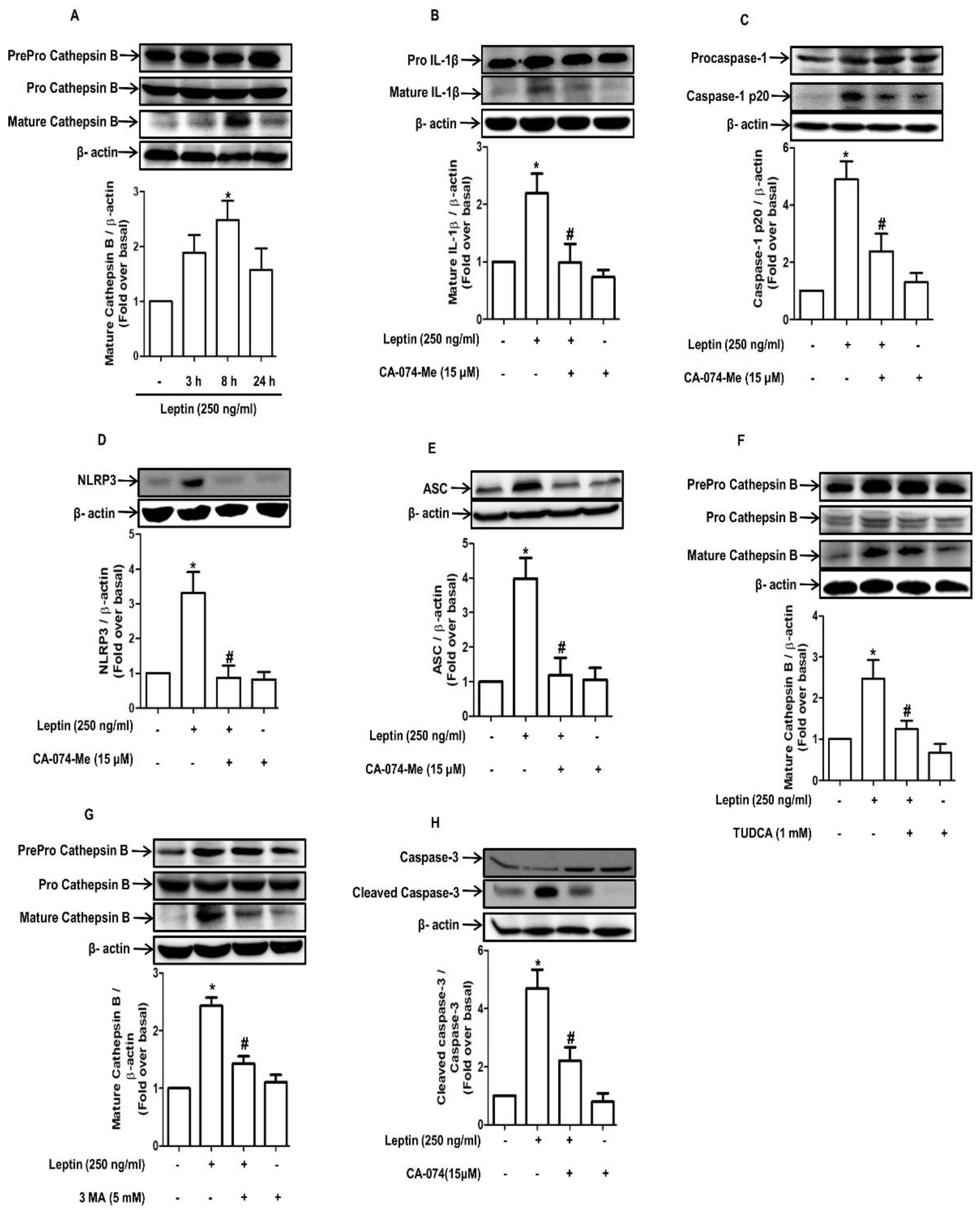
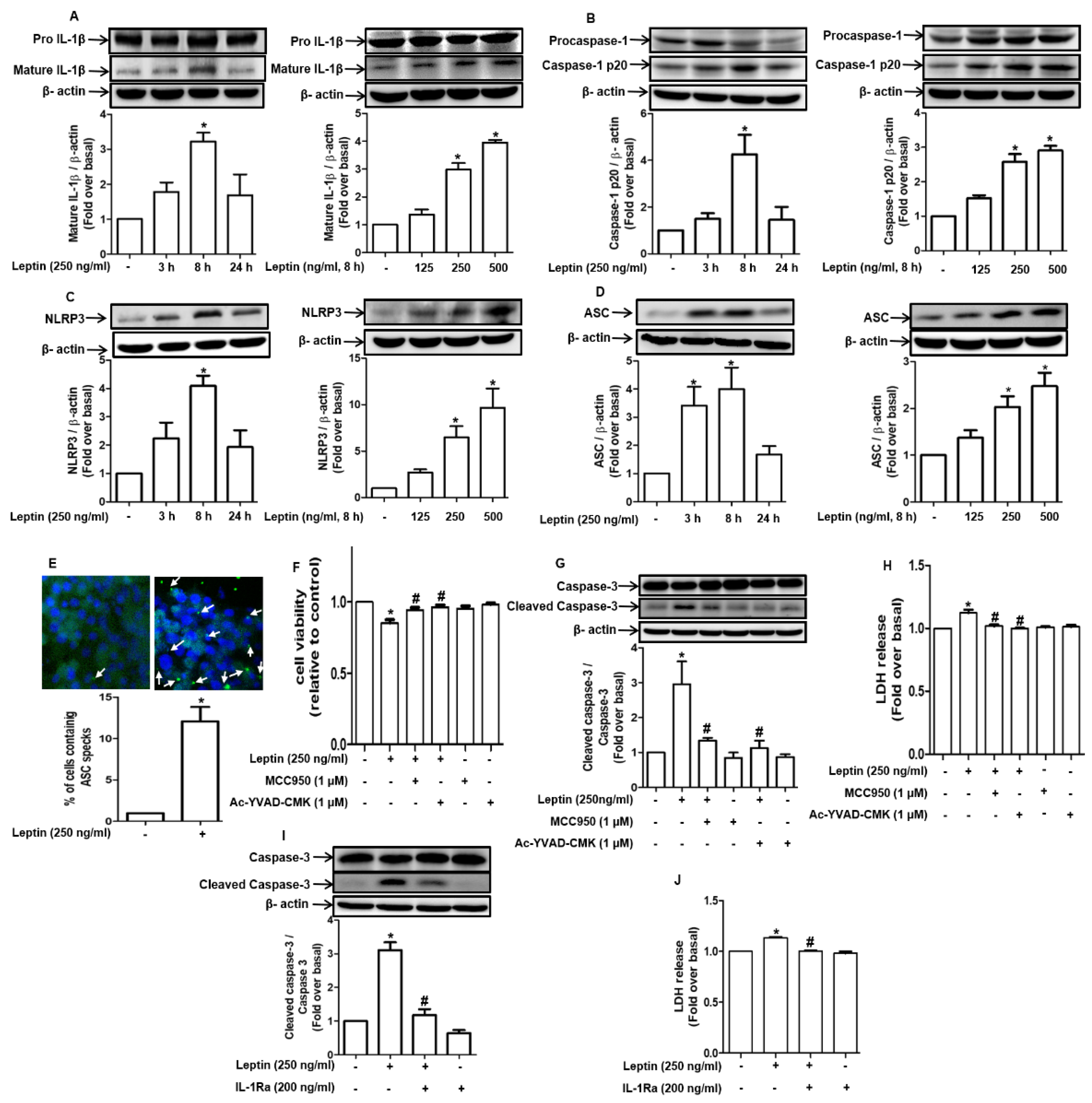
2.2. Cytotoxic Effect of Leptin in Hepatocytes Is Mediated via Inflammasome Activation
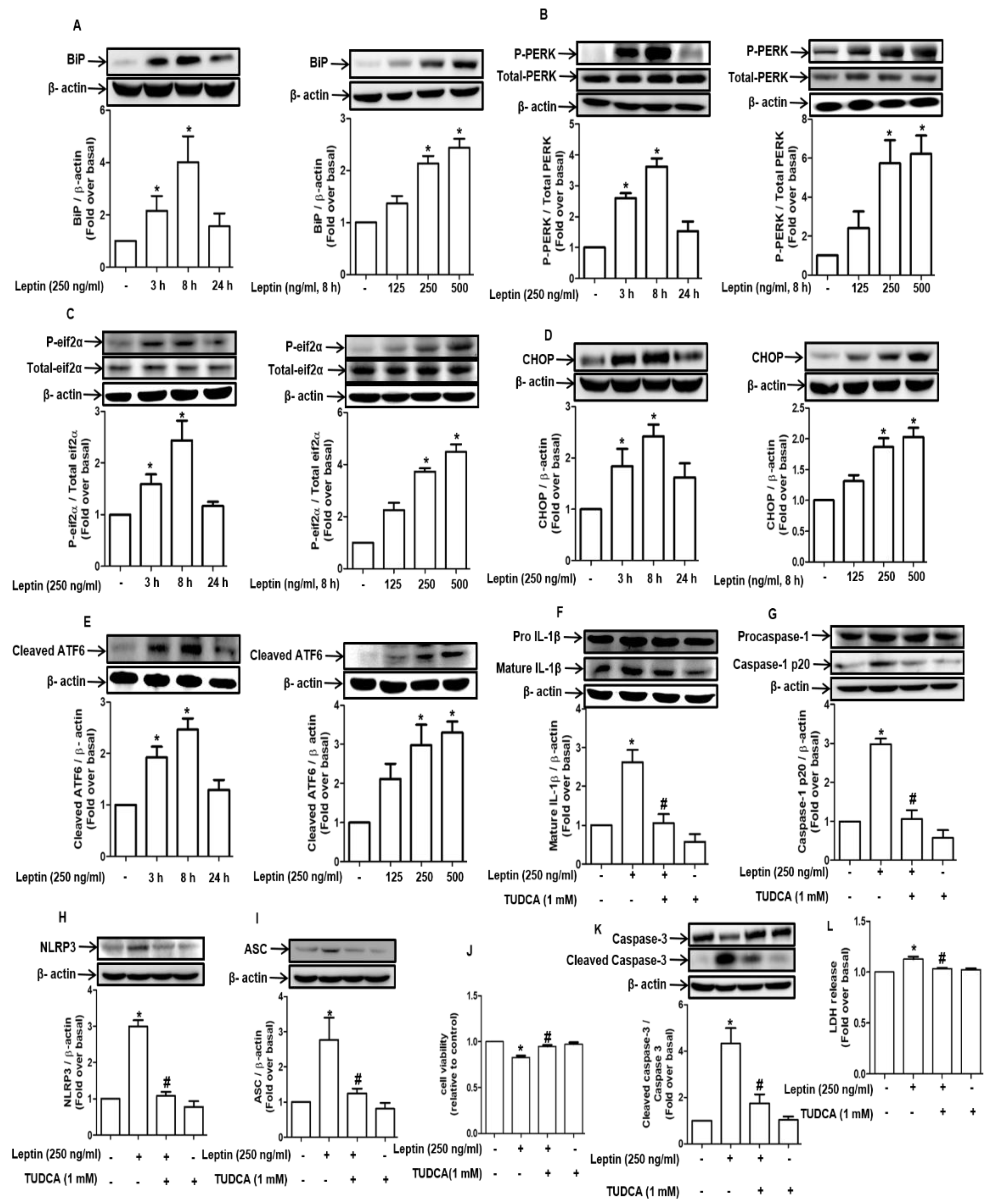
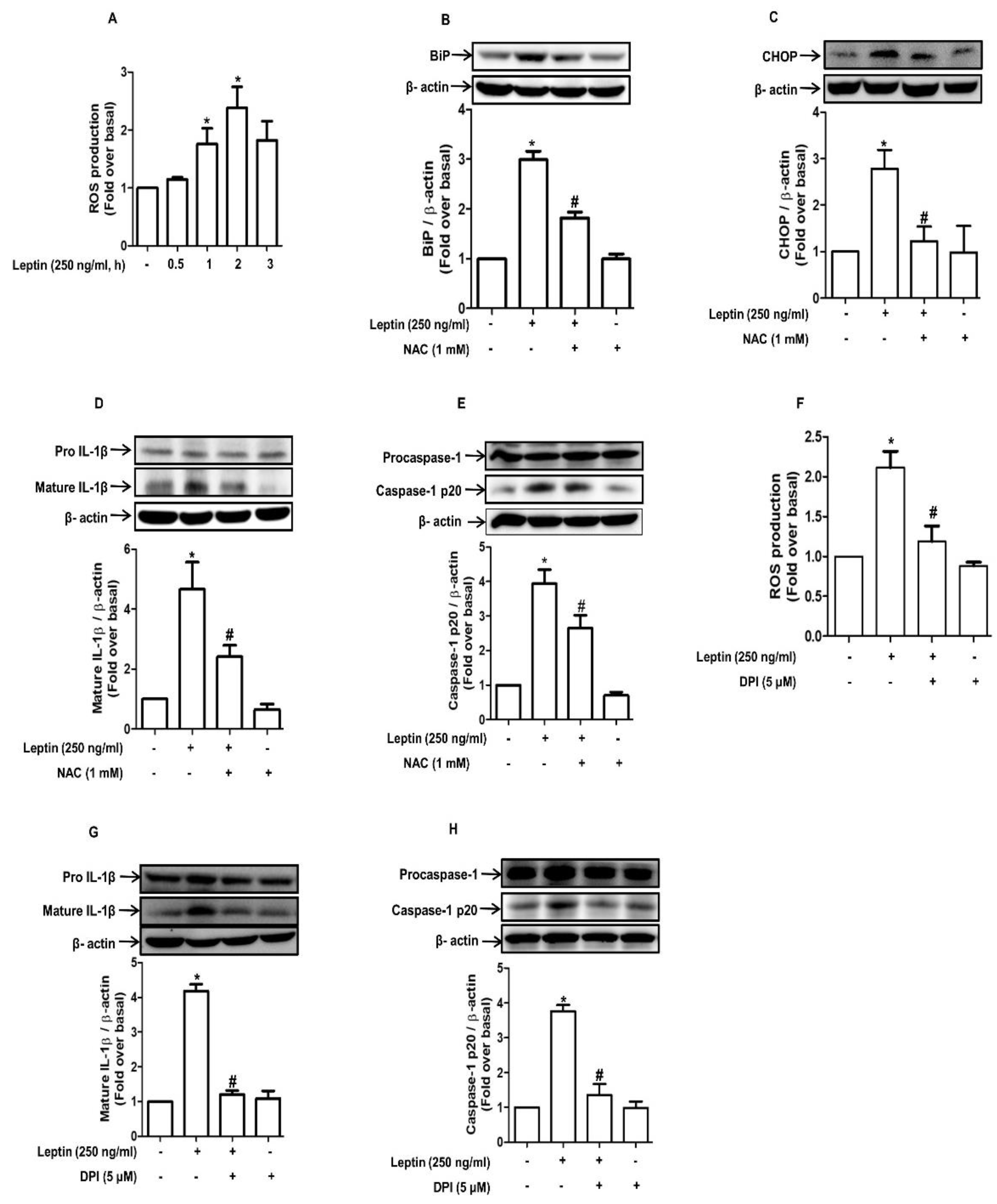
2.3. Inflammasome Activation by Leptin Is Mediated through ER Stress in Rat Hepatocytes
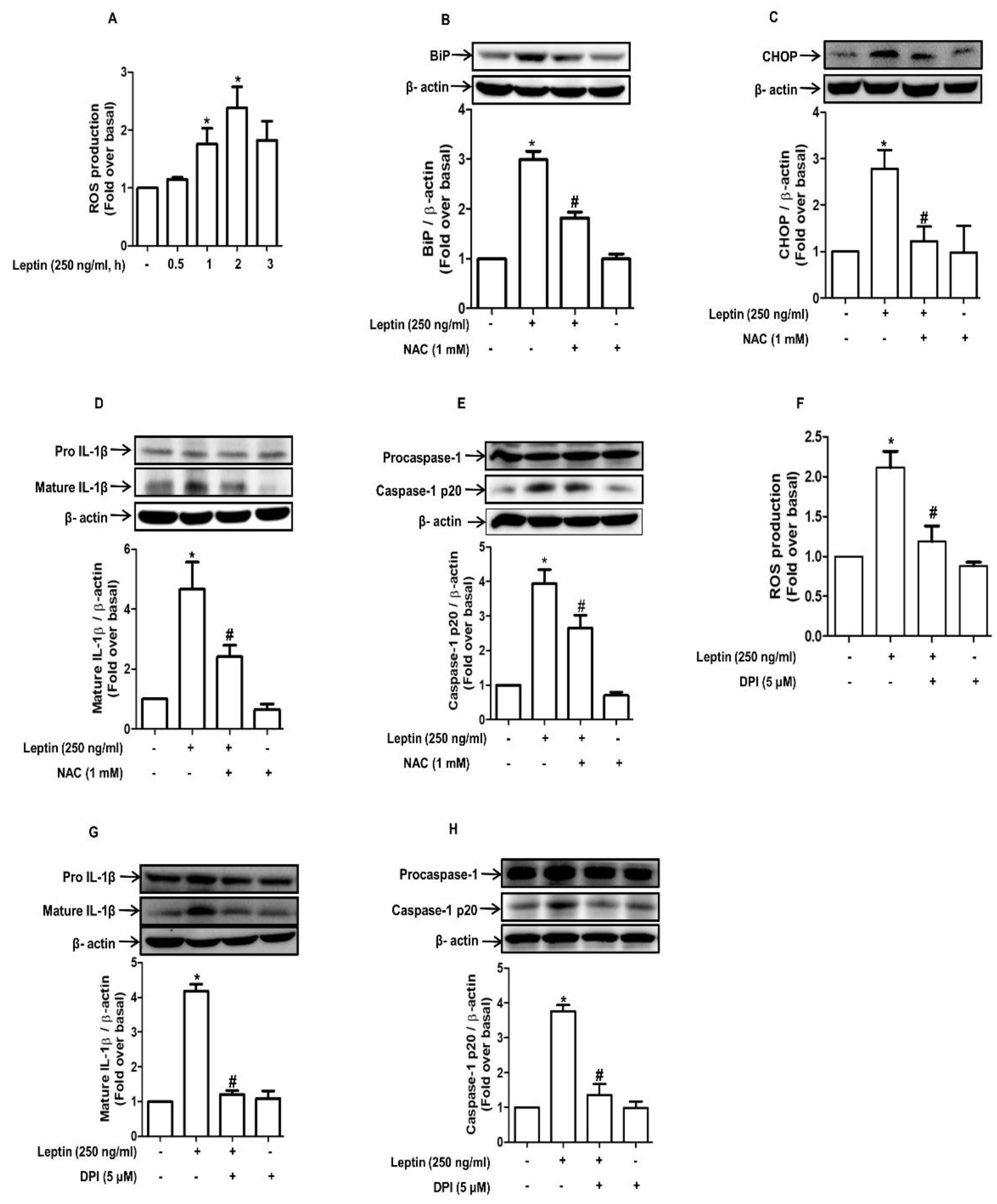
2.4. ROS Production Contributes to Leptin-Induced ER Stress and Inflammasome Activation in Rat Hepatocytes
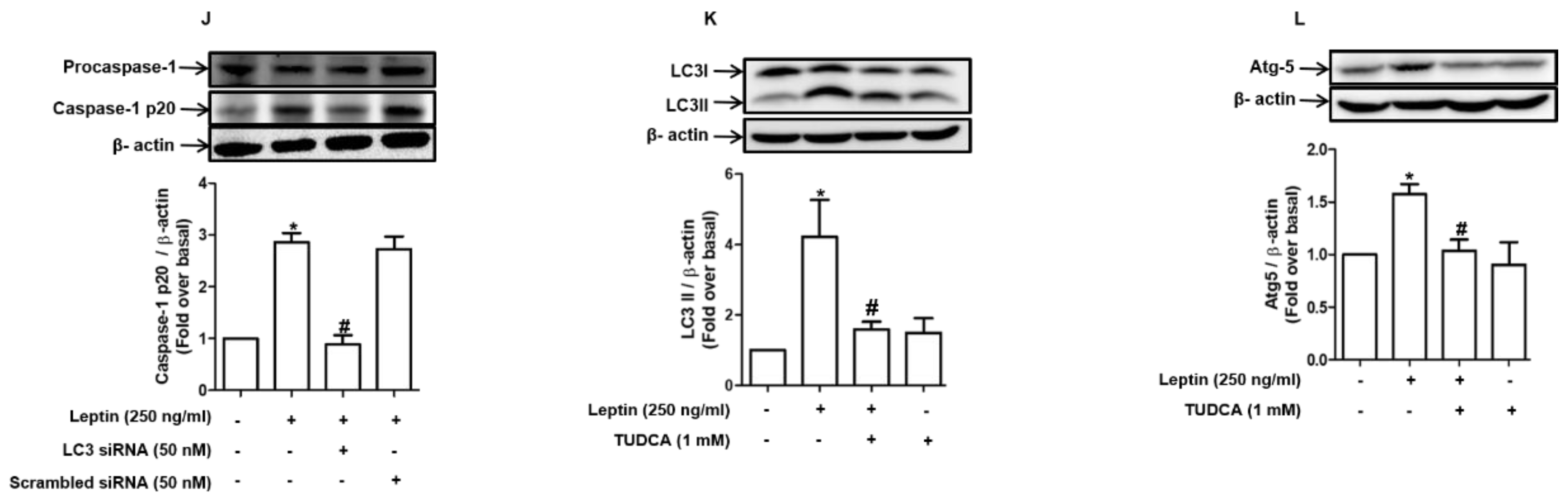
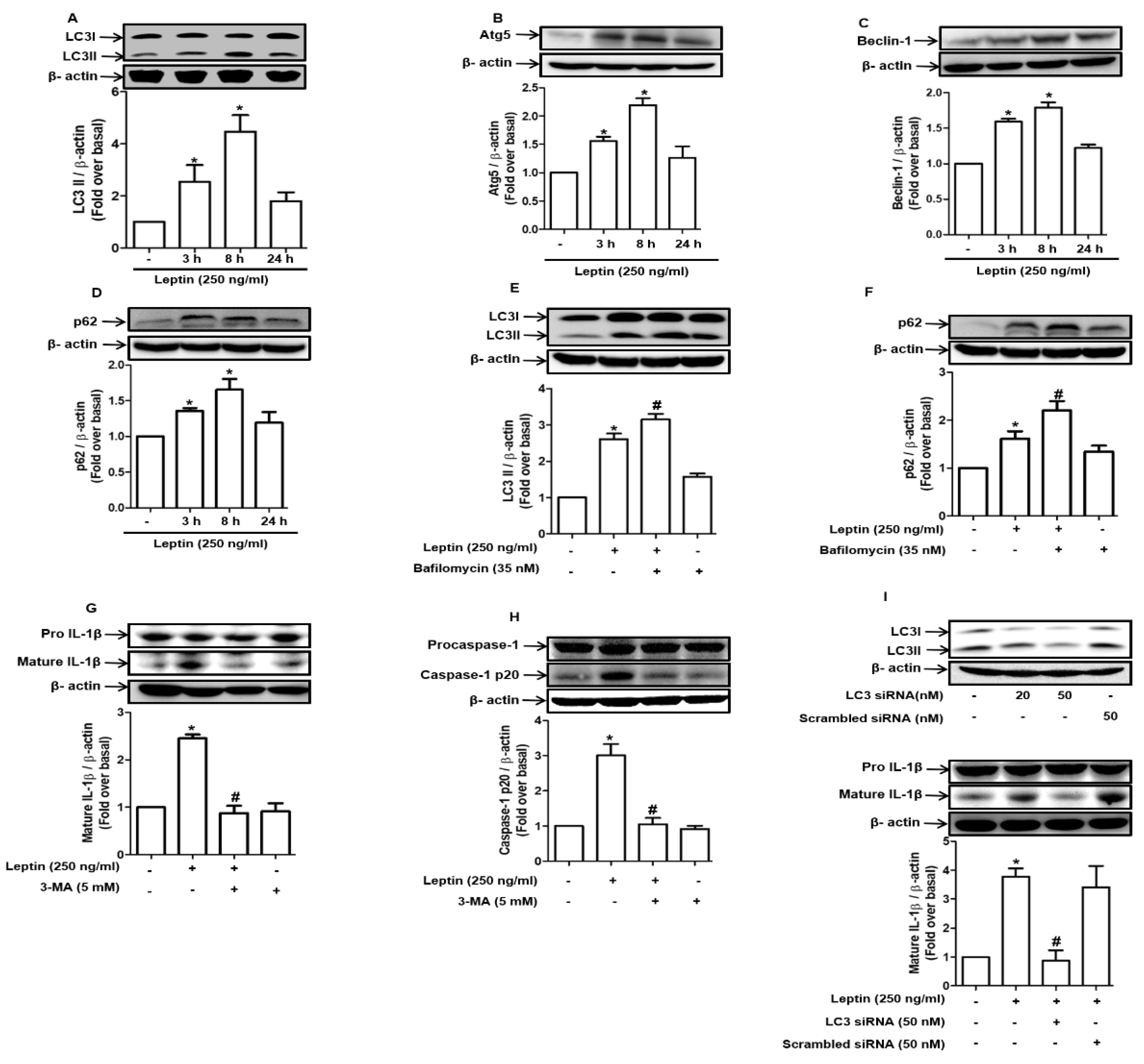
2.5. Autophagy Induction Mediates Inflammasome Activation by Leptin in Rat Hepatocytes
2.6. Maturation of Cathepsin B Induced by Autophagy Promotes Inflammasome Activation in Rat Hepatocytes
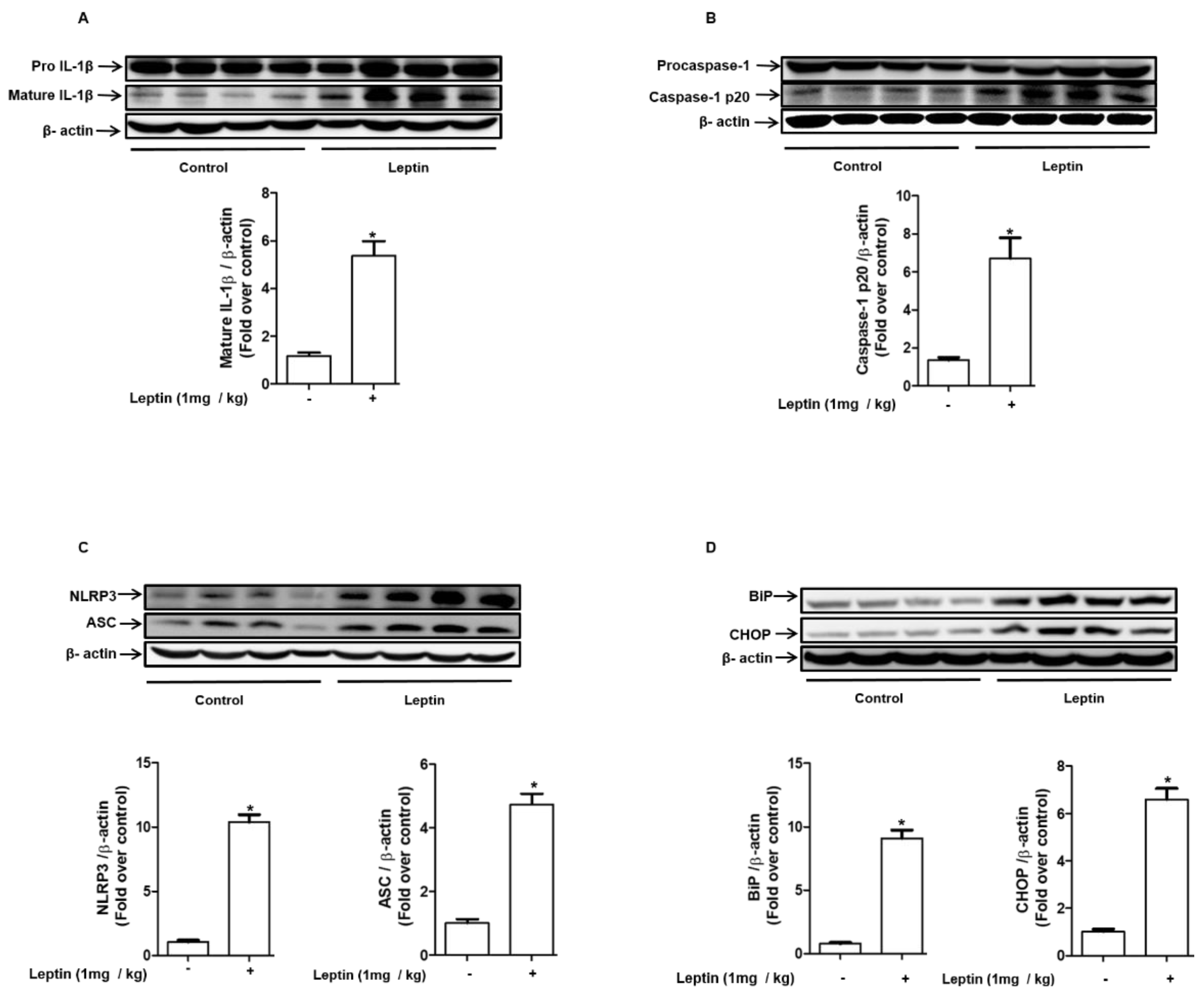
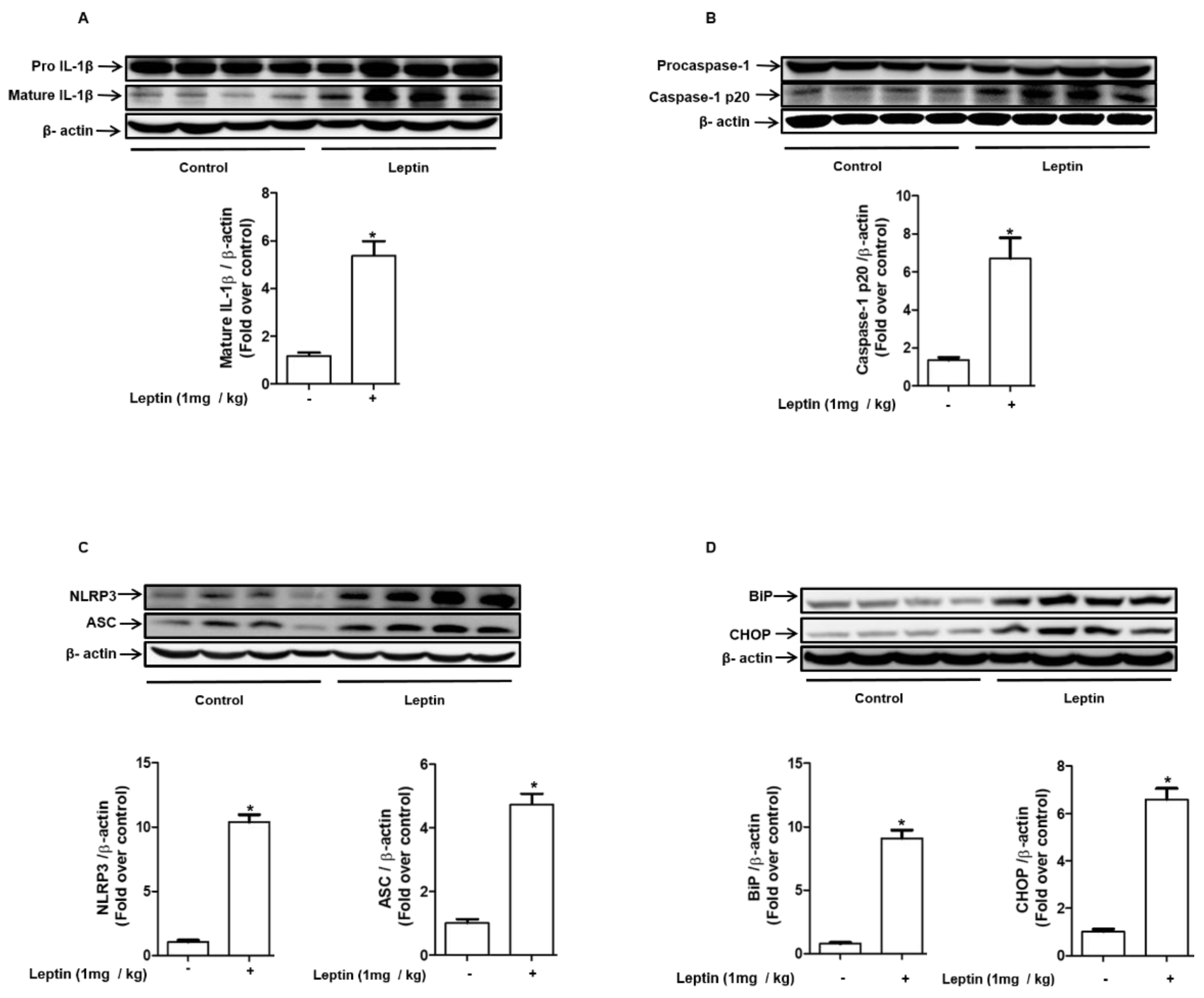
2.7. Leptin Induces Inflammasome Activation and ER Stress in Rat Liver
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isolation and Culture of Hepatocytes
4.3. Measurement of Cell Viability
4.4. Preparation of Cellular Extracts and Western Blot Analysis
4.5. Measurement of Reactive Oxygen Species (ROS) Production
4.6. Measurement of Lactate Dehydrogenase (LDH) Release
4.7. Immunocytochemistry for the Detection of ASC Speck
4.8. Transient Transfection with Small Interfering RNA (siRNA)
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsai, J.P. The association of serum leptin levels with metabolic diseases. Tzu-Chi Med J. 2017, 29, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.Y.; Kim, D.H.; Chun, K.H. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab. Anim. Res. 2018, 34, 133–139. [Google Scholar] [CrossRef]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, G.L.; Schrum, J.E.; De Nardo, C.M.; Latz, E. Intracellular sensing of microbes and danger signals by the inflammasomes. Immunol. Rev. 2011, 243, 119–135. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Park, P.H. Globular adiponectin protects rat hepatocytes against acetaminophen-induced cell death via modulation of the inflammasome activation and ER stress: Critical role of autophagy induction. Biochem. Pharmacol. 2018, 154, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Bamboat, Z.M.; Balachandran, V.P.; Ocuin, L.M.; Obaid, H.; Plitas, G.; DeMatteo, R.P. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010, 51, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrin, K.A.; Flach, R.J.R.; DiStefano, M.T.; Matevossian, A.; Friedline, R.H.; Jung, D.; Kim, J.K.; Czech, M.P. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS ONE 2014, 9, e107265. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Kang, T.B. The Molecular Links between Cell Death and Inflammasome. Cells 2019, 8, 1057. [Google Scholar] [CrossRef] [Green Version]
- Raut, P.K.; Kim, S.H.; Choi, D.Y.; Jeong, G.S.; Park, P.H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef]
- Lopez, S.; Rodriguez-Gallardo, S.; Sabido-Bozo, S.; Muñiz, M. Endoplasmic Reticulum Export of GPI-Anchored Proteins. Int. J. Mol. Sci. 2019, 20, 3506. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Lin, H.; Wen, J.; Sun, Q.; Gao, Y.; Xu, X.; Wang, J.; Zhang, J.; Weng, D. The kinase receptor-interacting protein 1 is required for inflammasome activation induced by endoplasmic reticulum stress. Cell Death Dis. 2018, 9, 641. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Schweichel, J.U.; Merker, H.J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–266. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compan, V.; Martin-Sanchez, F.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrin, P. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J. Immunol. 2015, 194, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Du, L.; Zhang, L.; Hu, Y.; Xia, W.; Wu, J.; Zhu, J.; Chen, L.; Zhu, F.; Li, C.; et al. Cathepsin B contributes to autophagy-related 7 (Atg7)-induced nod-like receptor 3 (NLRP3)-dependent proinflammatory response and aggravates lipotoxicity in rat insulinoma cell line. J. Biol. Chem. 2013, 288, 30094–30104. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef]
- Steer, S.A.; Scarim, A.L.; Chambers, K.T.; Corbett, J.A. Interleukin-1 stimulates beta-cell necrosis and release of the immunological adjuvant HMGB1. PLoS Med. 2006, 3, e17. [Google Scholar] [CrossRef]
- Shen, J.; Xu, S.; Zhou, H.; Liu, H.; Jiang, W.; Hao, J.; Hu, Z. IL-1β induces apoptosis and autophagy via mitochondria pathway in human degenerative nucleus pulposus cells. Sci. Rep. 2017, 7, 41067. [Google Scholar] [CrossRef]
- Wang, X.-J.; Kong, K.-M.; Qi, W.-L.; Ye, W.-L.; Song, P.-S. Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacol. Sin. 2005, 26, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Ammendrup, A.; Maillard, A.; Nielsen, K.; Andersen, N.A.; Serup, P.; Madsen, O.D.; Mandrup-Poulsen, T.; Bonny, C. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000, 49, 1468–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Kearney, J.; Shanahan, K.A.; Hams, E.; Sugisawa, R.; Connolly, D.; Doran, C.G.; Muñoz-Wolf, N.; Gürtler, C.; Fitzgerald, K.A.; et al. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity 2019, 50, 1412–1424.e1416. [Google Scholar] [CrossRef]
- Pham, D.-V.; Park, P.-H. Recent insights on modulation of inflammasomes by adipokines: A critical event for the pathogenesis of obesity and metabolism-associated diseases. Arch. pharmacal Res. 2020, 43, 997–1016. [Google Scholar] [CrossRef]
- Liao, Y.; Hussain, T.; Liu, C.; Cui, Y.; Wang, J.; Yao, J.; Chen, H.; Song, Y.; Sabir, N.; Hussain, M.; et al. Endoplasmic Reticulum Stress Induces Macrophages to Produce IL-1β During Mycobacterium bovis Infection via a Positive Feedback Loop Between Mitochondrial Damage and Inflammasome Activation. Front. Immunol. 2019, 10, 268. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef] [Green Version]
- Katiyar, S.; Liu, E.; Knutzen, C.A.; Lang, E.S.; Lombardo, C.R.; Sankar, S.; Toth, J.I.; Petroski, M.D.; Ronai, Z.; Chiang, G.G. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep. 2009, 10, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Zhai, W.; Lu, X.; Wang, G. The Cross-Links of Endoplasmic Reticulum Stress, Autophagy, and Neurodegeneration in Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 691881. [Google Scholar] [CrossRef]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Lee, J.C. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. USA 2015, 112, 9322–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchard, M.; Mignot, G.; Derangère, V.; Chalmin, F.; Chevriaux, A.; Végran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Schmidt, F.I.; Ploegh, H.L. Editorial: Crystal death: It’s not always the inflammasome. J. Leukoc. Biol. 2017, 102, 1–4. [Google Scholar] [CrossRef]
- Han, H.; Li, J.; Feng, X.; Zhou, H.; Guo, S.; Zhou, W. Autophagy-related genes are induced by histone deacetylase inhibitor suberoylanilide hydroxamic acid via the activation of cathepsin B in human breast cancer cells. Oncotarget 2017, 8, 53352–53365. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; He, Y.; Ye, X.; Cai, Y.; Xu, J.; Zhang, L.; Li, M.; Liu, H.; Wang, S.; Xia, Z. Activation of autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome activation and attenuates myocardial ischemia-reperfusion injury in diabetic rats. J. Diabetes Investig. 2020, 11, 1126–1136. [Google Scholar] [CrossRef]
- Mora, R.; Régnier-Vigouroux, A. Autophagy-driven cell fate decision maker: Activated microglia induce specific death of glioma cells by a blockade of basal autophagic flux and secondary apoptosis/necrosis. Autophagy 2009, 5, 419–421. [Google Scholar] [CrossRef] [Green Version]
- Khakurel, A.; Park, P.H. Globular adiponectin protects hepatocytes from tunicamycin-induced cell death via modulation of the inflammasome and heme oxygenase-1 induction. Pharmacol. Res. 2018, 128, 231–243. [Google Scholar] [CrossRef]
- Pham, D.V.; Raut, P.K.; Pandit, M.; Chang, J.H.; Katila, N.; Choi, D.Y.; Jeong, J.H.; Park, P.H. Globular Adiponectin Inhibits Breast Cancer Cell Growth through Modulation of Inflammasome Activation: Critical Role of Sestrin2 and AMPK Signaling. Cancers 2020, 12, 613. [Google Scholar] [CrossRef] [Green Version]
- Pun, N.T.; Park, P.H. Adiponectin inhibits inflammatory cytokines production by Beclin-1 phosphorylation and B-cell lymphoma 2 mRNA destabilization: Role for autophagy induction. Br. J. Pharmacol. 2018, 175, 1066–1084. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baral, A.; Park, P.-H. Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. Int. J. Mol. Sci. 2021, 22, 12589. https://doi.org/10.3390/ijms222212589
Baral A, Park P-H. Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. International Journal of Molecular Sciences. 2021; 22(22):12589. https://doi.org/10.3390/ijms222212589
Chicago/Turabian StyleBaral, Ananda, and Pil-Hoon Park. 2021. "Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes" International Journal of Molecular Sciences 22, no. 22: 12589. https://doi.org/10.3390/ijms222212589
APA StyleBaral, A., & Park, P.-H. (2021). Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. International Journal of Molecular Sciences, 22(22), 12589. https://doi.org/10.3390/ijms222212589