
On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis



Abstract
:1. Introduction
2. Results and Discussion
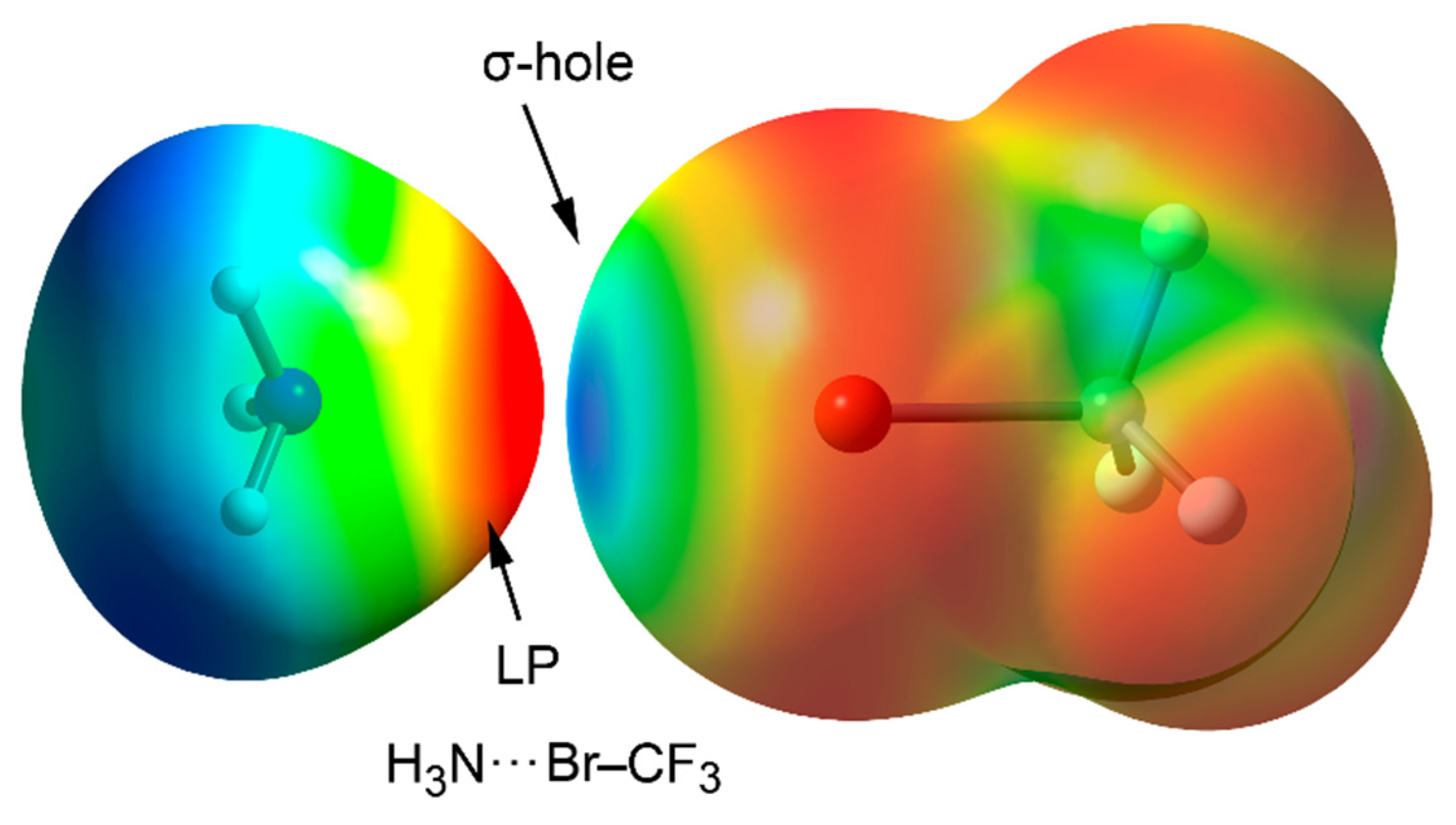
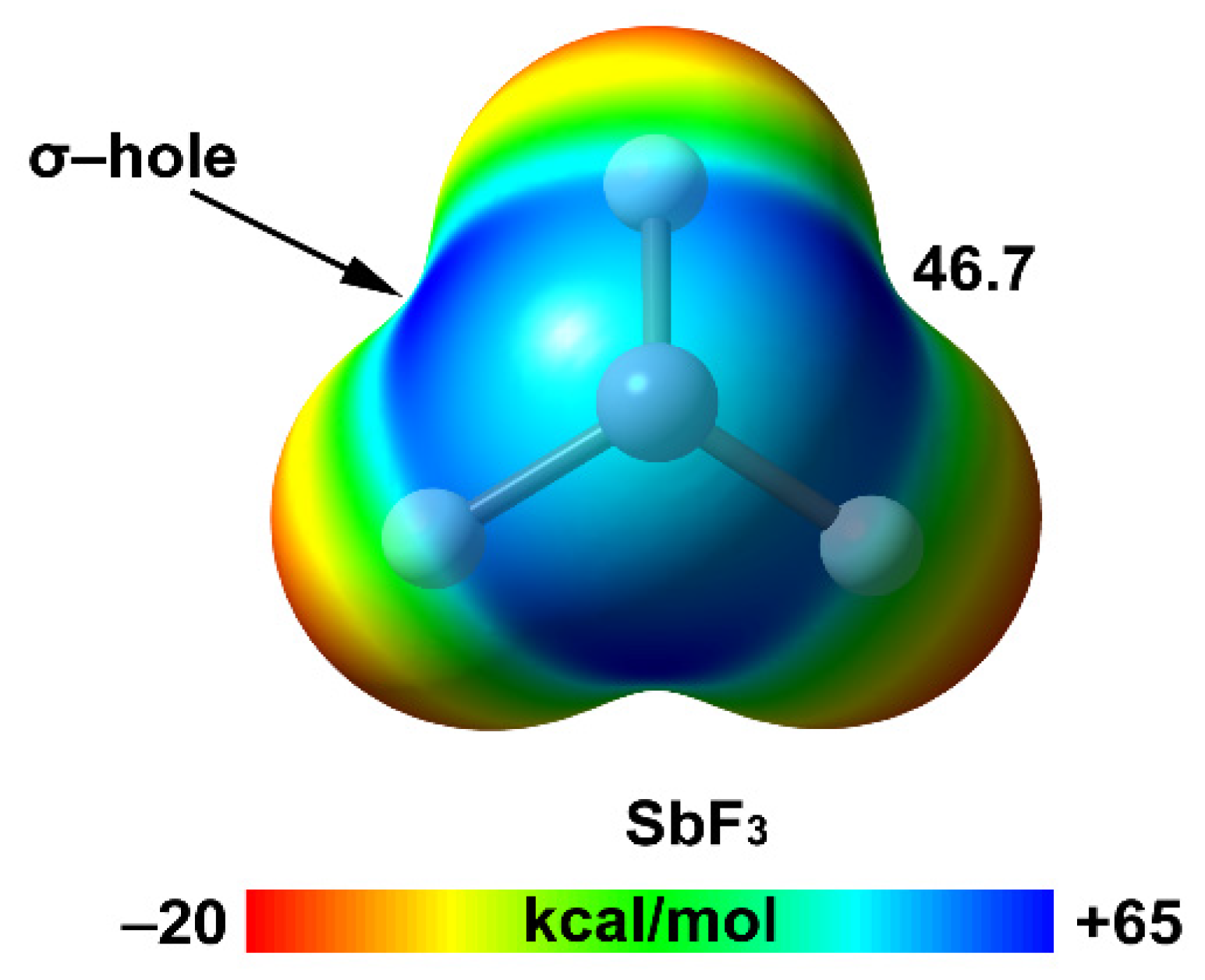
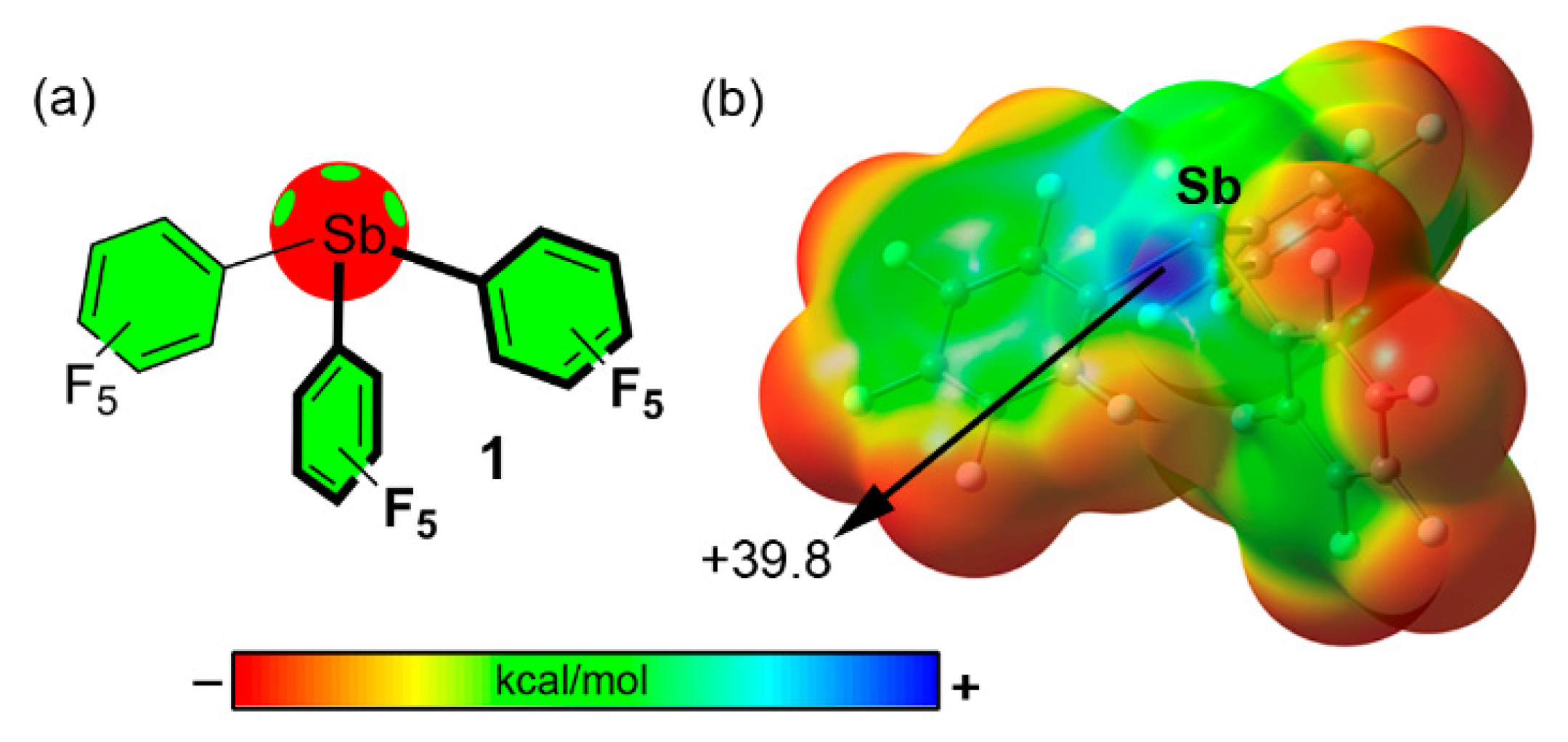
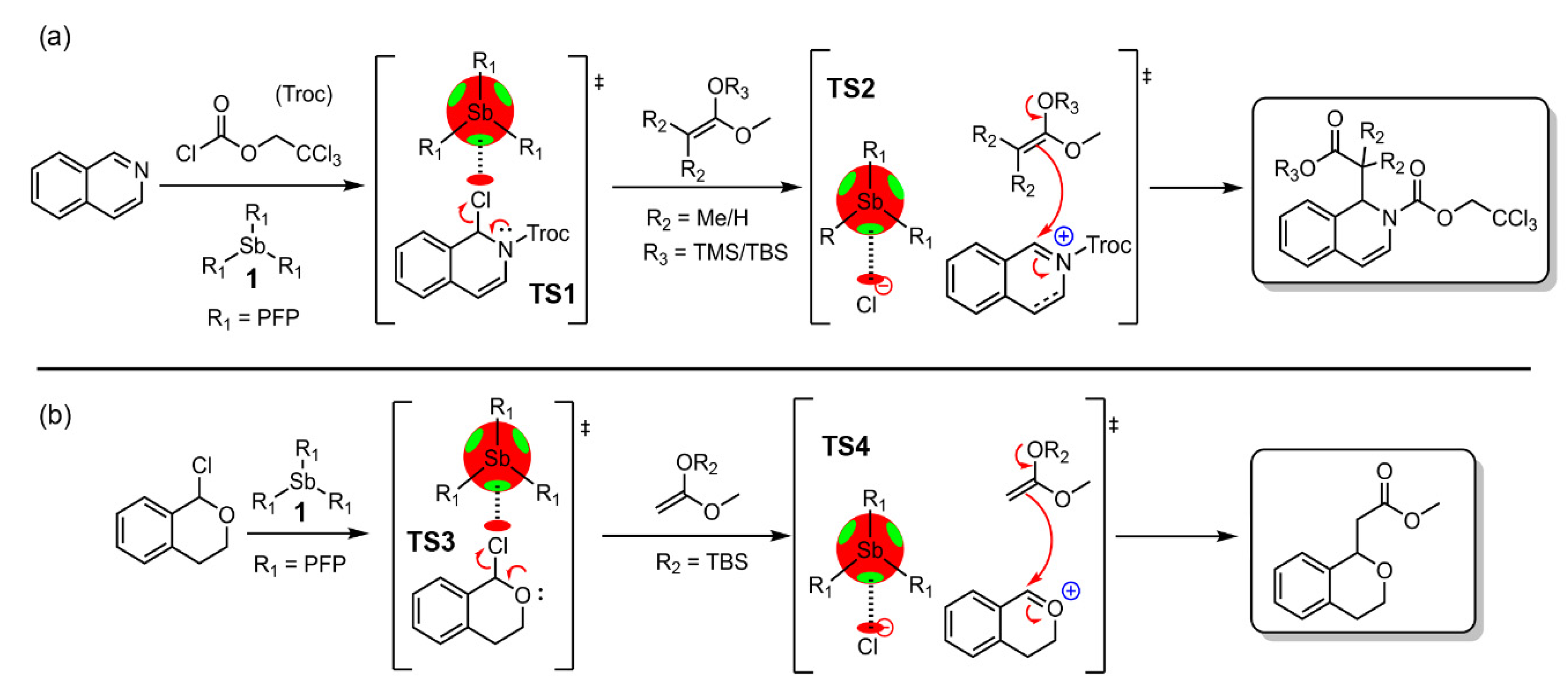
2.1. Pnictogen Bonding
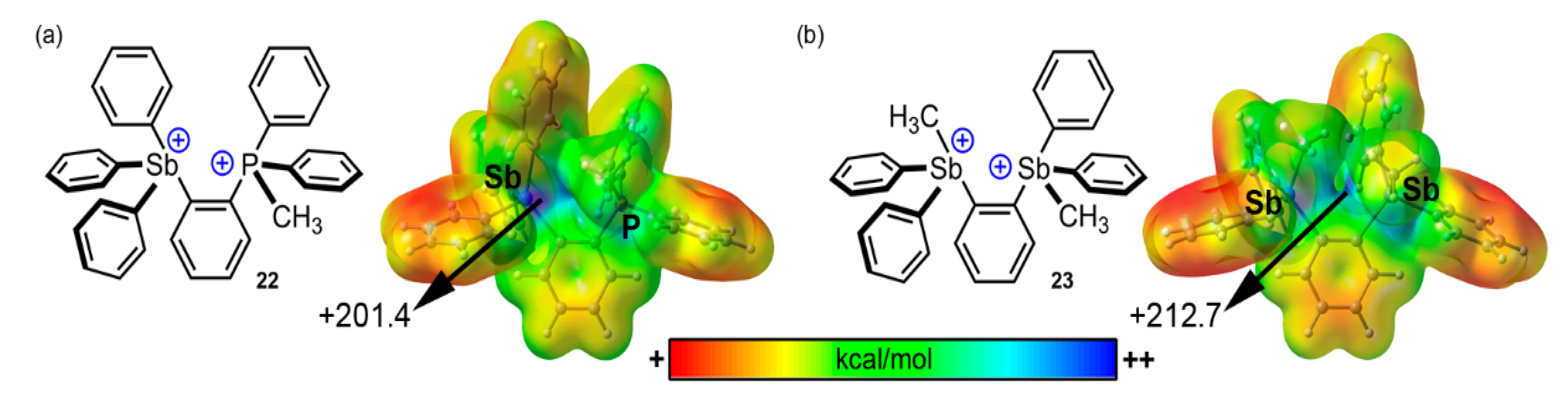
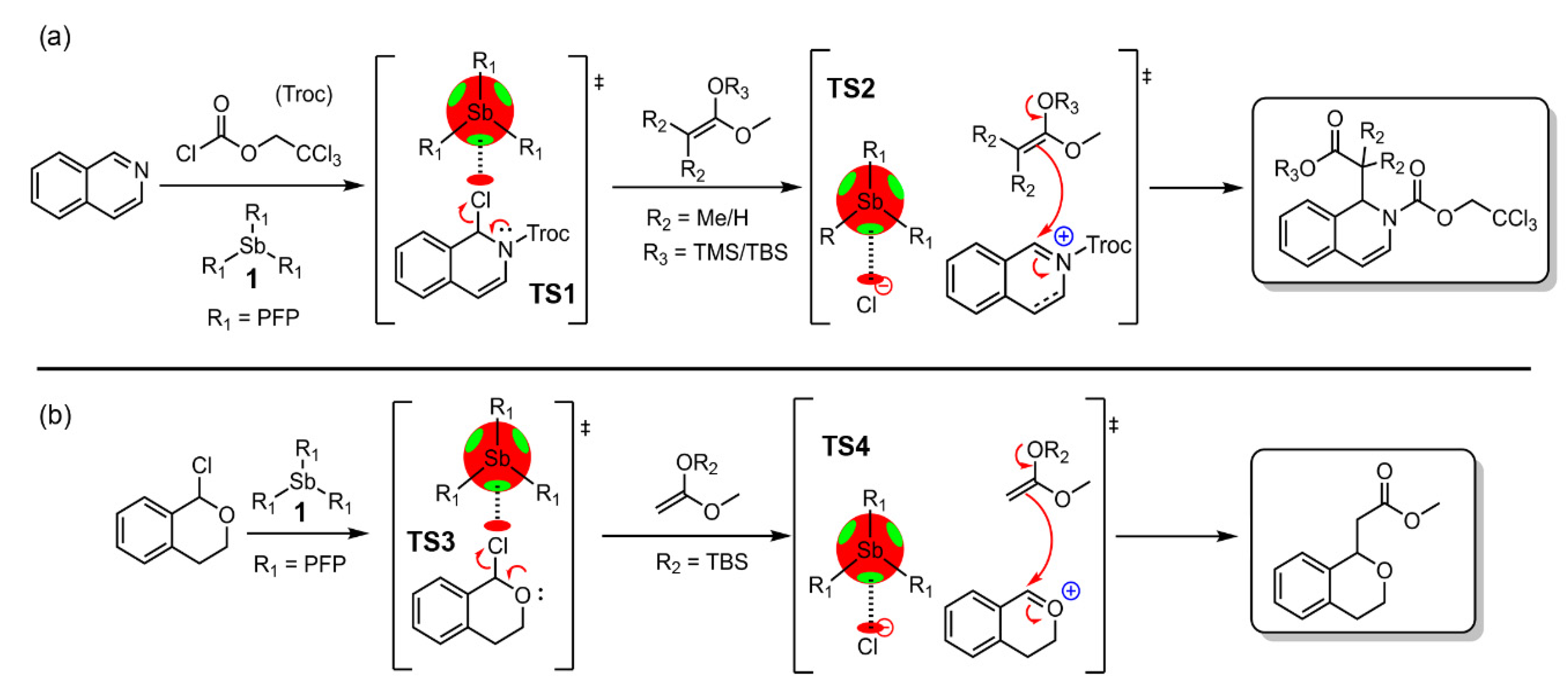
Arsenic and Antimony
2.2. Chalcogen Bonding
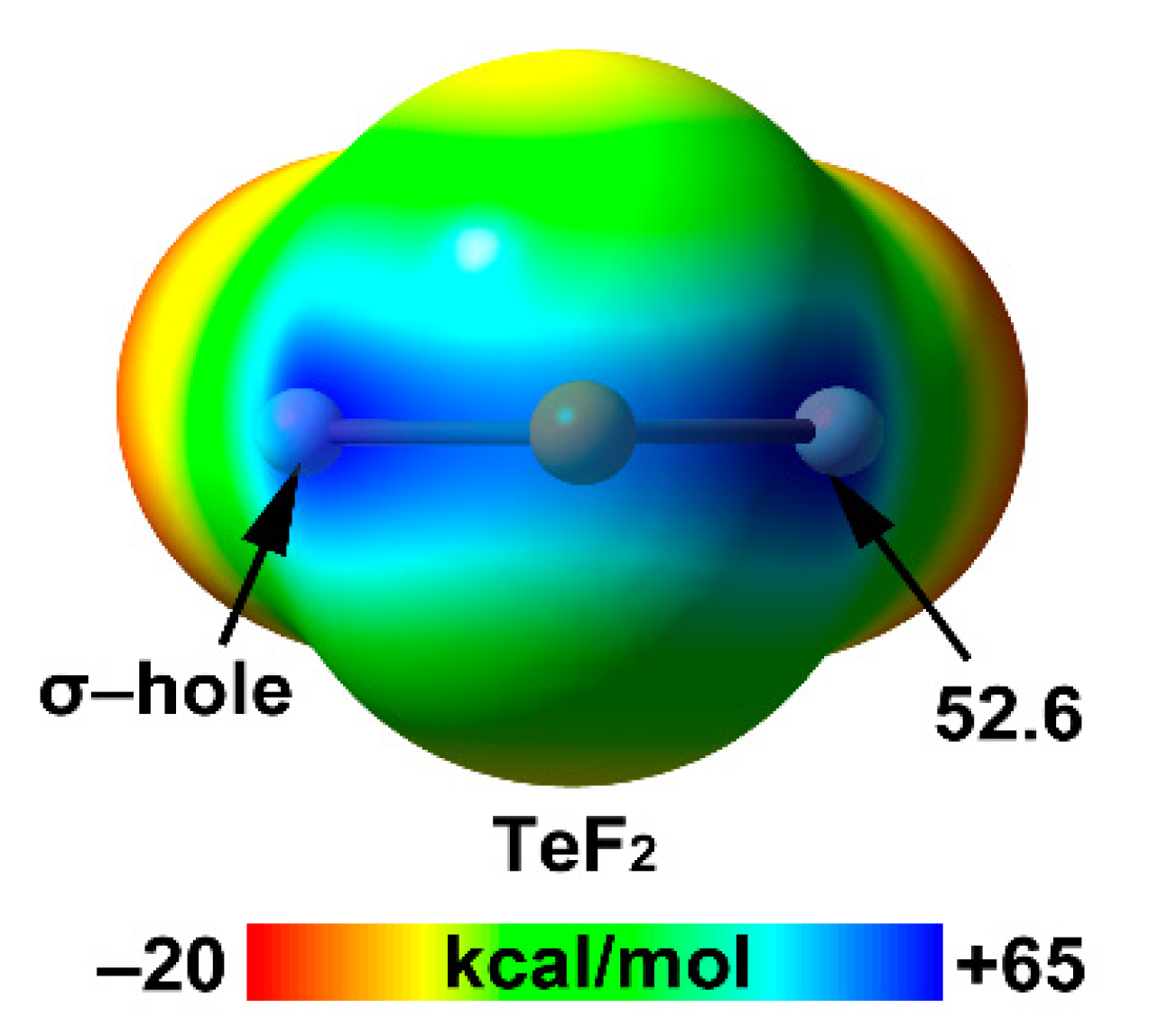
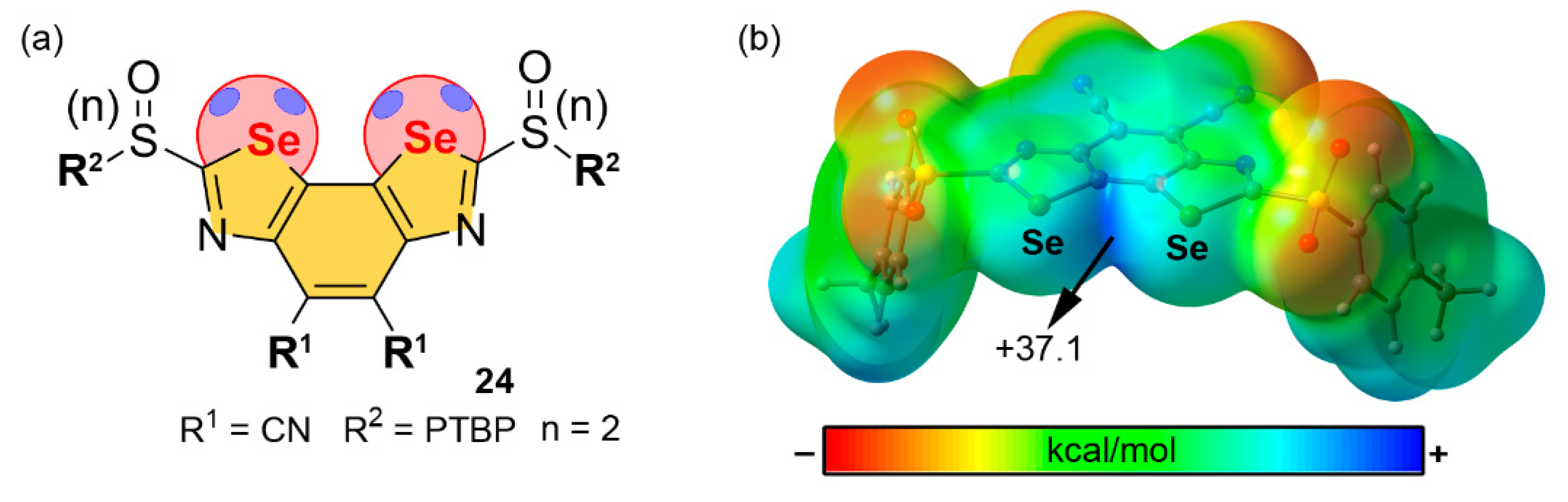
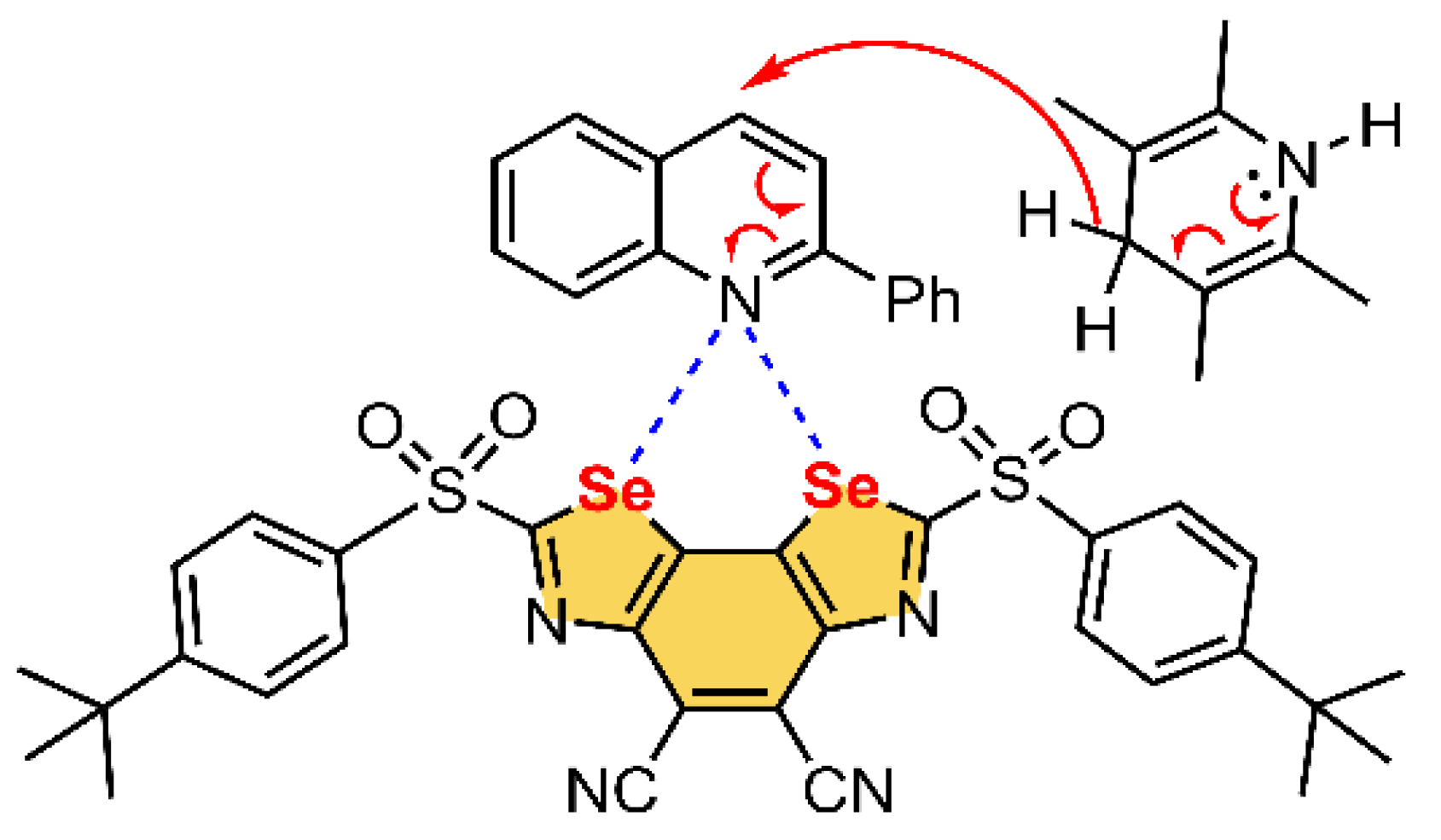
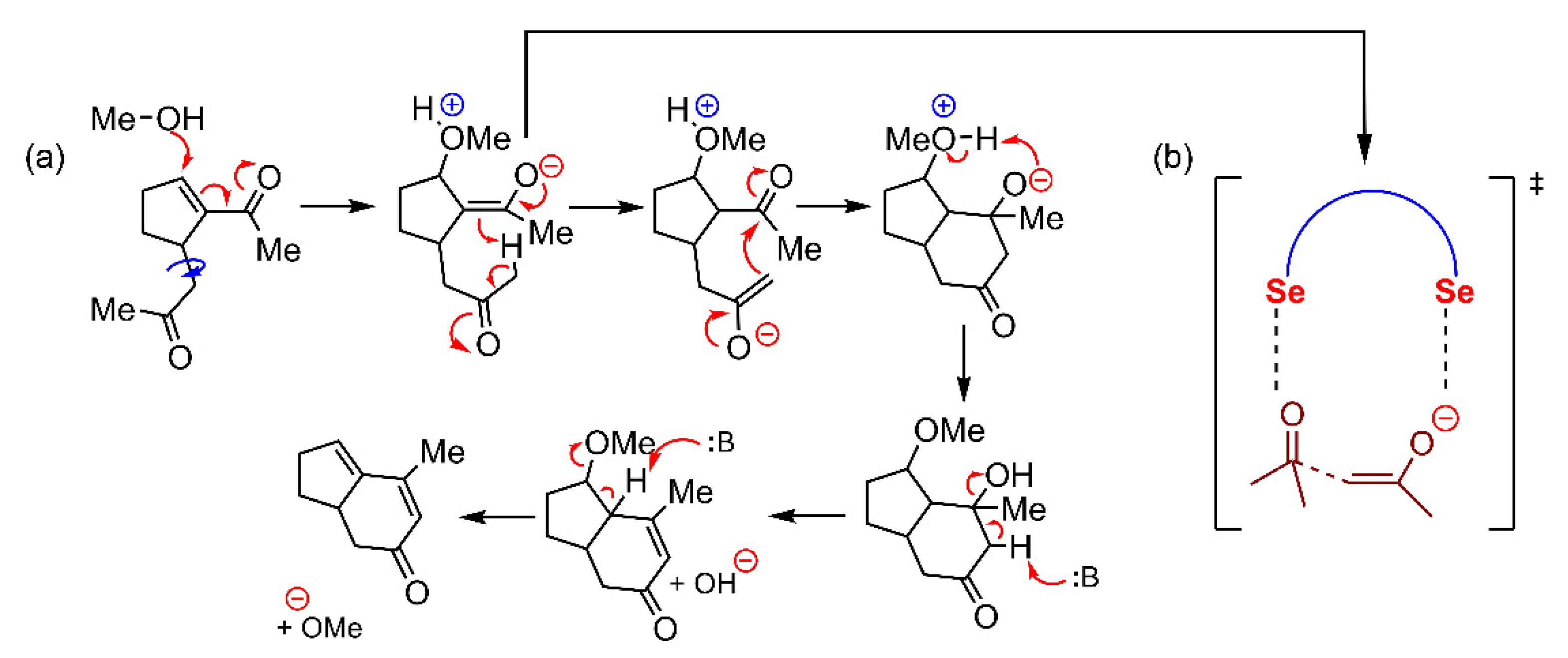
Selenium and Tellurium
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pizzi, A.; Pigliacelli, C.; Bergamaschi, G.; Gori, A.; Metrangolo, P. Biomimetic engineering of the molecular recognition and self-assembly of peptides and proteins via halogenation. Coord. Chem. Rev. 2020, 411, 213242. [Google Scholar] [CrossRef]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C(sp3) atoms as tetrel bond donors: A crystallographic survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Von der Heiden, D.; Vanderkooy, A.; Erdélyi, M. Halogen bonding in solution: NMR spectroscopic approaches. Coord. Chem. Rev. 2020, 407, 213147. [Google Scholar] [CrossRef]
- Gill, H.; Gokel, M.R.; McKeever, M.; Negin, S.; Patel, M.B.; Yin, S.; Gokel, G.W. Supramolecular pore formation as an antimicrobial strategy. Coord. Chem. Rev. 2020, 412, 213264. [Google Scholar] [CrossRef]
- Xu, Y.; Szell, P.M.J.; Kumar, V.; Bryce, D.L. Solid-state NMR spectroscopy for the analysis of element-based non-covalent interactions. Coord. Chem. Rev. 2020, 411, 213237. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Jin, W.J. Halogen bonding in room-temperature phosphorescent materials. Coord. Chem. Rev. 2020, 404, 213107. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel bond and coordination of triel centres—Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171. [Google Scholar] [CrossRef]
- Fromm, K.M. Chemistry of alkaline earth metals: It is not all ionic and definitely not boring! Coord. Chem. Rev. 2020, 408, 213192. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Scheiner, S.; Michalczyk, M.; Zierkiewicz, W. Coordination of anions by noncovalently bonded σ-hole ligands. Coord. Chem. Rev. 2020, 405, 213136. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. σ/π-Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming interactions from the electrophilic site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Terraneo, G.; Resnati, G. Bonding Matters. Cryst. Growth Des. 2017, 17, 1439–1440. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Daolio, A.; Pizzi, A.; Terraneo, G.; Frontera, A.; Resnati, G. σ-Holes Allow for Attractive Anion···anion Interactions Involving Perrhenate, Permanganate, and Pertechnetate Anions. ChemPhysChem 2021, 22, accepted. [Google Scholar] [CrossRef]
- Daolio, A.; Pizzi, A.; Calabrese, M.; Terraneo, G.; Bordignon, S.; Frontera, A.; Resnati, G. Molecular Electrostatic Potential and Noncovalent Interactions in Derivatives of Group 8 Elements. Angew. Chem. Int. Ed. 2021, 60, 20723–20727. [Google Scholar] [CrossRef]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.H.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef] [Green Version]
- Daolio, A.; Pizzi, A.; Terraneo, G.; Ursini, M.; Frontera, A.; Resnati, G. Anion⋅⋅⋅Anion Coinage Bonds: The Case of Tetrachloridoaurate. Angew. Chem. Int. Ed. 2021, 60, 14385–14389. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauzá, A. Regium–π bonds: An Unexplored Link between Noble Metal Nanoparticles and Aromatic Surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef]
- Terrón, A.; Buils, A.; Mooibroek, T.J.; Barceló, M.; García-Raso, A.; Fiol, J.J.; Frontera, A. Synthesis, X-ray characterization and regium bonding interactions of a trichlorido(1-hexylcytosine)gold(III) complex. Chem. Commun. 2020, 56, 3524–3527. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. (Eds.) Halogen Bonding: Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Beale, T.M.; Chudzinski, M.G.; Sarwar, M.G.; Taylor, M.S. Halogen bonding in solution: Thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. (Eds.) Halogen Bonding II Impact on Materials Chemistry and Life Sciences; Springer International Publishing AG: Heidelberg, Germany, 2015. [Google Scholar]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [Green Version]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From noncovalent chalcogen–chalcogen interactions to supramolecular aggregates: Experiments and calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The pnicogen bond in review: Structures, binding energies, bonding properties, and spin–spin coupling constants of complexes stabilized by pnicogen bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Heidelberg, Germany, 2015; pp. 191–264. [Google Scholar]
- Brammer, L. Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: Origins, current status and discussion. Faraday Discuss. 2017, 203, 485–507. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Lane, P. σ-Hole bonding and hydrogen bonding: Competitive interactions. Int. J. Quantum Chem. 2007, 107, 3046–3052. [Google Scholar] [CrossRef]
- Zhu, S.; Xing, C.; Xu, W.; Jin, G.; Li, Z. Halogen bonding and hydrogen bonding coexist in driving self-assembly process. Cryst. Growth Des. 2004, 4, 53–56. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Forni, A.; Gorynsztejn-Leben, M.; Kaivola, M.; Metrangolo, P.; Milani, R.; Shishido, A.; Pilati, T.; Resnati, G.; et al. Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly and Performance of Light-Responsive Supramolecular Polymers. Adv. Funct. Mater. 2012, 22, 2572–2579. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen bonds in crystal engineering: Like hydrogen bonds yet different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Forty years of progress in the study of the hydrogen bond. Struct. Chem. 2019, 30, 1119–1128. [Google Scholar] [CrossRef]
- Corradi, E.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G. Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly Processes. Angew. Chem. Int. Ed. 2000, 39, 1782–1786. [Google Scholar] [CrossRef]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The origin of chalcogen-bonding interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef] [Green Version]
- Bora, P.L.; Novák, M.; Novotný, J.; Foroutan-Nejad, C.; Marek, R. Supramolecular covalence in bifurcated chalcogen bonding. Chem. Eur. J. 2017, 23, 7315–7323. [Google Scholar] [CrossRef]
- Bhandary, S.; Sirohiwal, A.; Kadu, R.; Kumar, S.; Chopra, D. Dispersion stabilized Se/Te··· π double chalcogen bonding synthons in in situ cryocrystallized divalent organochalcogen liquids. Cryst. Growth Des. 2018, 18, 3734–3739. [Google Scholar] [CrossRef]
- Ams, M.R.; Trapp, N.; Schwab, A.; Milić, J.V.; Diederich, F. Chalcogen Bonding “2S–2N Squares” versus Competing Interactions: Exploring the Recognition Properties of Sulfur. Chem. Eur. J. 2019, 25, 323–333. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Concha, M.C. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Vargas-Baca, I.; Mansour, S.; Mahmoudkhani, A.H. The nature of the supramolecular association of 1, 2, 5-chalcogenadiazoles. J. Am. Chem. Soc. 2005, 127, 3184–3190. [Google Scholar] [CrossRef]
- De Vleeschouwer, F.; Denayer, M.; Pinter, B.; Geerlings, P.; De Proft, F. Characterization of chalcogen bonding interactions via an in-depth conceptual quantum chemical analysis. J. Comput. Chem. 2018, 39, 557–572. [Google Scholar] [CrossRef]
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Köppel, H. Theoretical Investigations on Heteronuclear Chalcogen− Chalcogen Interactions: On the Nature of Weak Bonds between Chalcogen Centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef]
- Bleiholder, C.; Werz, D.B.; Köppel, H.; Gleiter, R. Theoretical investigations on chalcogen− chalcogen interactions: What makes these nonbonded interactions bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An overview of strengths and directionalities of noncovalent interactions: σ-holes and π-holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Gomila, R.M.; Mooibroek, T.J.; Frontera, A. In A combined theoretical and CSD perspective on σ-hole interactions with tetrels, pnictogens, chalcogens, halogens, and noble gases. In Hot Topics in Crystal Engineering; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular complexes between silicon derivatives and electron-rich groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Fruchier, A.; Macquarrie, D.J.; Virgili, A. Aminopropylsilanes versus silatranes: An experimental and theoretical study. J. Organomet. Chem. 2001, 625, 148–153. [Google Scholar] [CrossRef]
- Grabowski, S.J. Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach. Crystals 2017, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Scilabra, P.; Kumar, V.; Ursini, M.; Resnati, G. Close contacts involving germanium and tin in crystal structures: Experimental evidence of tetrel bonds. J. Mol. Model. 2018, 24, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Lukevics, E.; Arsenyan, P.; Belyakov, S.; Popelis, J.; Pudova, O. Cycloaddition reactions of nitrile oxides to silyl-and germyl-substituted thiophene-1, 1-dioxides. Organometallics 1999, 18, 3187–3193. [Google Scholar] [CrossRef]
- Villaescusa, L.A.; Lightfoot, P.; Morris, R.E. Synthesis and structure of fluoride-containing GeO2 analogues of zeolite double four-ring building units. Chem. Commun. 2002, 2220–2221. [Google Scholar] [CrossRef] [PubMed]
- Calogero, S.; Ganis, P.; Peruzzo, V.; Tagliavini, G.; Valle, G. X-ray and Mössbauer studies of tricyclohexyltin (IV) halides. The crystal structures of (cyclo-C6H11)3SnX (X= F, Br and I). J. Organomet. Chem. 1981, 220, 11–20. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Kuang, D.-Z.; Zhang, F.-X.; Feng, Y.-L.; Xu, Z.-F. CCDC 211279: Experimental Crystal Structure Determination. Wuji Huaxue Xuebao 2003, 19, 1109. [Google Scholar]
- Breugst, M.; Koening, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 2020, 5473–5487. [Google Scholar] [CrossRef]
- Breugst, M.; von der Heiden, D.; Schmauck, J. Novel Noncovalent Interactions in Catalysis: A Focus on Halogen, Chalcogen, and Anion-π Bonding. Synthesis 2017, 49, 3224–3236. [Google Scholar] [CrossRef] [Green Version]
- Liao, L.; Zhao, X. Modern Organoselenium Catalysis: Opportunities and Challenges. Synlett 2021, 32, 1262–1268. [Google Scholar]
- Trujillo, C.; Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Halogen, chalcogen and pnictogen interactions in (XNO2)2 homodimers (X= F, Cl, Br, I). New J. Chem. 2015, 39, 6791–6802. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Grabowski, S.J. Pnicogen and hydrogen bonds: Complexes between PH3X+ and PH2X systems. Phys. Chem. Chem. Phys. 2015, 17, 3261–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esrafili, M.D.; Mohammadian-Sabet, F.; Baneshi, M.M. The dual role of halogen, chalcogen, and pnictogen atoms as Lewis acid and base: Triangular XBr:SHX:PH2X complexes (X = F, Cl, Br, CN, NC, OH, NH2, and OCH3). Int. J. Quantum Chem. 2015, 115, 1580–1586. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Bauzá, A.; Quinonero, D.; Deyà, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined Cambridge structural database and theoretical study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. Pnicogen Bonds between X–PH3 (X = O, S, NH, CH2) and Phosphorus and Nitrogen Bases. J. Phys. Chem. A 2014, 118, 1527–1537. [Google Scholar] [CrossRef] [Green Version]
- Shukla, R.; Chopra, D. Characterization of N⋯ O non-covalent interactions involving σ-holes: “electrostatics” or “dispersion”. Phys. Chem. Chem. Phys. 2016, 18, 29946–29954. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S.D. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Scilabra, P.; Terraneo, G.; Resnati, G. Fluorinated elements of Group 15 as pnictogen bond donor sites. J. Fluor. Chem. 2017, 203, 62–74. [Google Scholar] [CrossRef]
- Mokrai, R.; Jaie Barrett, J.; Apperley, D.C.; Benkő, Z.; Heift, D. Tweaking the Charge Transfer: Bonding Analysis of Bismuth(III) Complexes with a Flexidentate Phosphane Ligand. Inorg. Chem. 2020, 59, 8916–8924. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Pavan, M.S.; Guru Row, T.N. Experimental validation of ’pnicogen bonding’ in nitrogen by charge density analysis. Phys. Chem. Chem. Phys. 2015, 17, 2330–2334. [Google Scholar] [CrossRef]
- Joshi, P.R.; Ramanathan, N.; Sundararajan, K.; Sankaran, K. Evidence for phosphorus bonding in phosphorus trichloride–methanol adduct: A matrix isolation infrared and ab initio computational study. J. Phys. Chem. A 2015, 119, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Korlyukov, A.A.; Lyssenko, K.A. Experimental charge density evidence for pnicogen bonding in a crystal of ammonium chloride. ChemPhysChem 2015, 16, 676–681. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Beer, P.D. Sigma-hole interactions in anion recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Hirai, M.; Cho, J.; Gabbaï, F. Promoting the Hydrosilylation of Benzaldehyde by Using a Dicationic Antimony-Based Lewis Acid: Evidence for the Double Electrophilic Activation of the Carbonyl Substrate. Chem. Eur. J. 2016, 22, 6537–6541. [Google Scholar] [CrossRef]
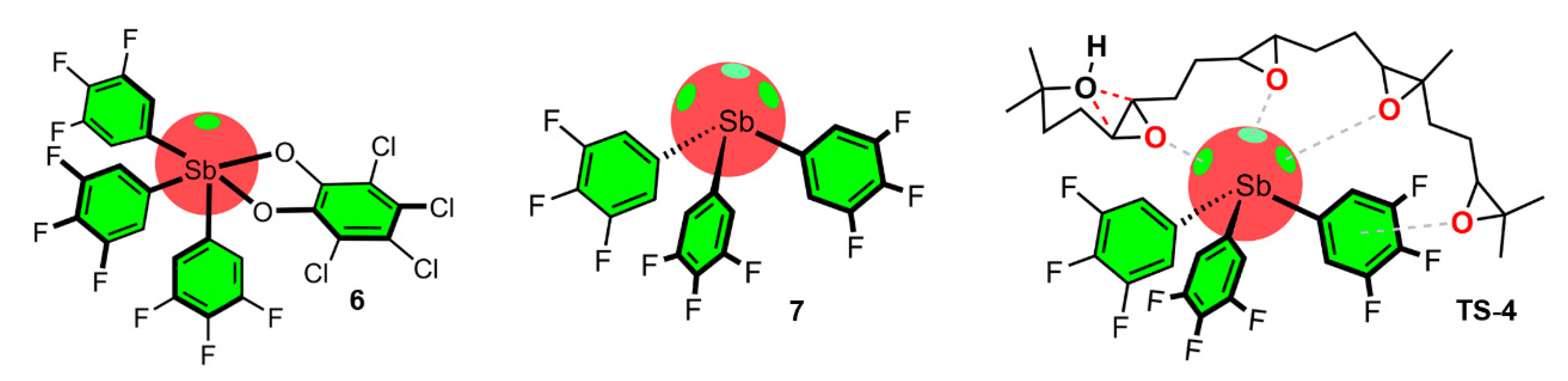
- Qiu, J.; Unruh, D.K.; Cozzolino, A.F. Design, Synthesis, and Structural Characterization of a Bisantimony(III) Compound for Anion Binding and the Density Functional Theory Evaluation of Halide Binding through Antimony Secondary Bonding Interactions. J. Phys. Chem. A 2016, 120, 9257–9269. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
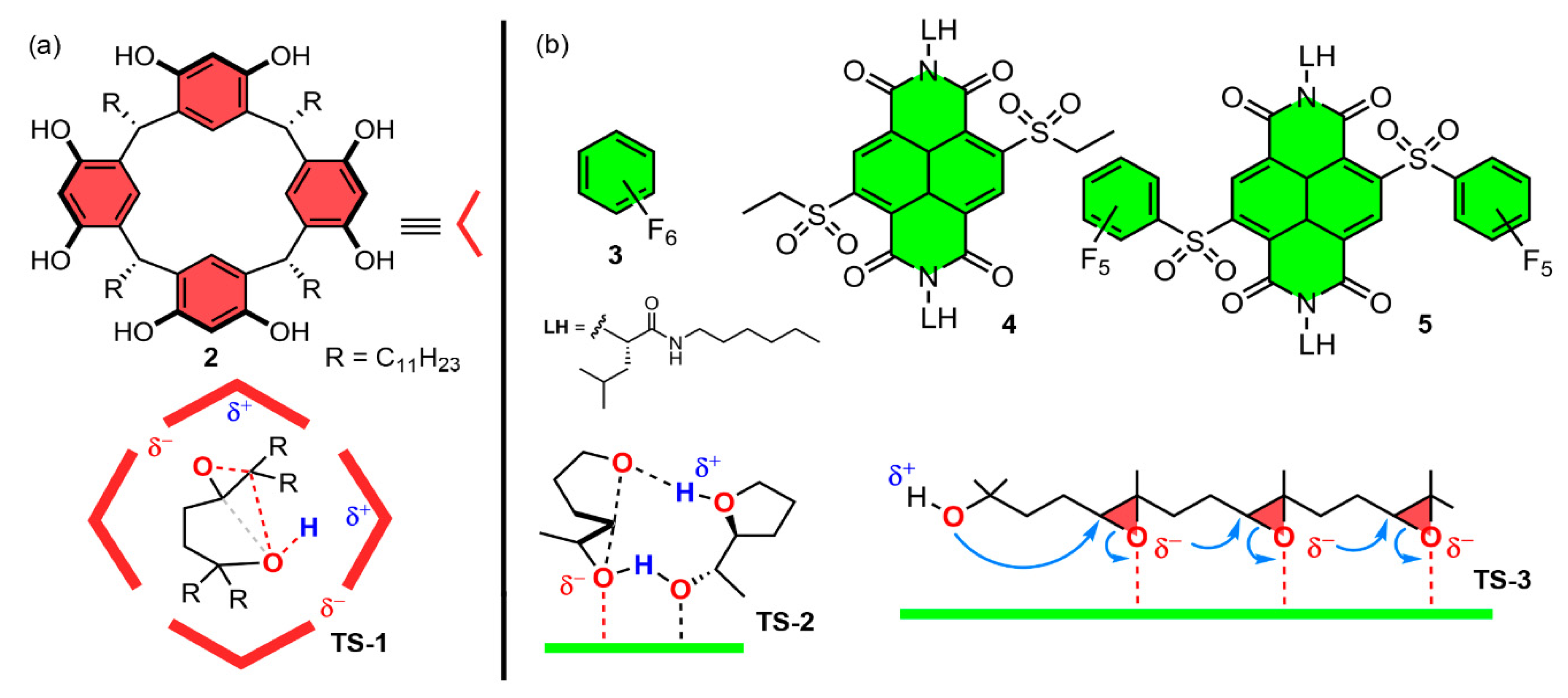
- Paraja, M.; Gini, A.; Sakai, N.; Matile, S. Pnictogen-Bonding Catalysis: An Interactive Tool to Uncover Unorthodox Mechanisms in Polyether Cascade Cyclizations. Chem. Eur. J. 2020, 26, 15471–15476. [Google Scholar] [CrossRef]
- Gini, A.; Paraja, A.; Galmés, B.; Besnard, C.; Poblador-Bahamonde, A.I.; Sakai, N.; Frontera, A.; Matile, S. Pnictogen-bonding catalysis: Brevetoxin-type polyether cyclizations. Chem. Sci. 2020, 11, 7086–7091. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Li, T.R.; Chen, H.; Gini, A.; Zhang, X.; Rosset, S.; Mazet, C.; Tiefenbacher, K.; Matile, S. Bioinspired Ether Cyclizations within a π‐Basic Capsule Compared to Autocatalysis on π‐Acidic Surfaces and Pnictogen‐Bonding Catalysts. Chem. Eur. J. 2021, 27, 12215–12223. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, J.; Ding, W.-Y.; Li, S.; Xiang, S.H.; Tan, B. Asymmetric Pnictogen-Bonding Catalysis: Transfer Hydrogenation by a Chiral Antimony(V) Cation/Anion Pair. J. Am. Chem. Soc. 2021, 143, 6382–6387. [Google Scholar] [CrossRef]
- Yang, M.; Hirai, M.; Gabbaï, F. Phosphonium-stilbonium and bis-stilbonium cations as pnictogen-bonding catalysts for the transfer hydrogenation of quinolines. Dalton Trans. 2019, 48, 6685–6689. [Google Scholar] [CrossRef]
- Humeniuk, H.V.; Gini, A.; Hao, X.; Coelho, F.; Sakai, N.; Matile, S. Pnictogen-Bonding Catalysis and Transport Combined: Polyether Transporters Made In Situ. J. Am. Chem. Soc. 2021, 1, 1588–1593. [Google Scholar] [CrossRef]
- Vermeeren, P.; Tiezza, M.D.; van Dongen, M.; Fernández, I.; Bickelhaupt, F.M.; Hamlin, T.A. Lewis Acid-Catalyzed Diels-Alder Reactions: Reactivity Trends across the Periodic Table. Chem. Eur. J. 2021, 27, 10610–10620. [Google Scholar] [CrossRef]
- Li, Y.; Meng, L.; Sun, C.; Zeng, Y. Organocatalysis by Halogen, Chalcogen, and Pnictogen Bond Donors in Halide Abstraction Reactions: An Alternative to Hydrogen Bond-Based Catalysis. J. Phys. Chem. A 2020, 124, 3815–3824. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lu, Y.; Zhu, Z.; Liu, H. Pnictogen, chalcogen, and halogen bonds in catalytic systems: Theoretical study and detailed comparison. J. Mol. Model. 2020, 26, 16. [Google Scholar] [CrossRef]
- Schafer, A.G.; Wieting, J.M.; Fisher, T.J.; Mattson, A.E. Chiral Silanediols in Anion-Binding Catalysis. Angew.Chem. Int. Ed. 2013, 52, 11321–11324. [Google Scholar] [CrossRef] [PubMed]
- Zurro, M.; Asmus, S.; Bamberger, J.; Beckendorf, S.; García Mancheño, O. Chiral Triazoles in Anion-Binding Catalysis: New Entry to Enantioselective Reissert-Type Reactions. Chem. Eur. J. 2016, 22, 3785–3793. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Kumatabara, Y.; Shimizu, S.; Maruoka, K.; Shirakawa, S. Hydrogen-bonding catalysis of sulfonium salts. Chem. Commun. 2017, 53, 119–122. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Garrett, G.E.; Gibson, G.L.; Straus, R.N.; Seferos, D.S.; Taylor, M.S. Chalcogen bonding in solution: Interactions of benzotelluradiazoles with anionic and uncharged Lewis bases. J. Am. Chem. Soc. 2015, 137, 4126–4133. [Google Scholar] [CrossRef]
- Navarro-García, E.; Galmés, B.; Velasco, M.D.; Frontera, A.; Caballero, A. Anion Recognition by Neutral Chalcogen Bonding Receptors: Experimental and Theoretical Investigations. Chem. Eur. J. 2020, 26, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, A.F.; Elder, P.J.W.; Vargas-Vaca, I. A survey of tellurium-centered secondary-bonding supramolecular synthons. Coord. Chem. Rev. 2011, 255, 1426–1438. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Nziko, V.P.N.; Scheiner, S. Chalcogen Bonding between Tetravalent SF4 and Amines. J. Phys. Chem. A 2014, 118, 10849–10856. [Google Scholar] [CrossRef]
- Benz, S.; Mareda, J.; Besnard, C.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds: Neutral benzodiselenazole scaffolds with high-precision selenium donors of variable strength. Chem. Sci. 2017, 8, 8164–8169. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, S.; Liu, Z.; Zhao, L.; Zhang, J.; Hao, J.; Wang, Y. Chalcogen-Chalcogen Bonding Catalysis Enables Assembly of Discrete Molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, H.; Feng, L.; Yu, Q.; Hao, J.; Zhu, R.; Wang, Y. Dual Chalcogen-Chalcogen Bonding Catalysis. J. Am. Chem. Soc. 2020, 142, 3117–3124. [Google Scholar] [CrossRef]
- Bao, L.; Kong, X.; Wang, Y. Noncovalent Chalcogen-Bonding Catalysis Using ppm-Level Catalyst Loading to Achieve Cyanosilylation of Ketones. Asian J. Org. Chem. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S.M. Chalcogen Bonding Catalysis of a Nitro-Michael Reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.; Aubert, E.; Pale, P.; Mamane, V. Chalcogen-Bonding Catalysis with Telluronium Cations. Angew. Chem. Int. Ed. 2021, 60, 19281–19286. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Steve Tse, Y.L.; Ke, Z.; Yeung, Y.Y. Bis-selenonium Cations as Bidentate Chalcogen Bond Donors in Catalysis. ACS Catal. 2021, 11, 12632–12642. [Google Scholar] [CrossRef]
- Zhou, B.; Gabbai, F.P. Lewis Acidic Telluronium Cations: Enhanced Chalcogen-Bond Donor Properties and Application to Transfer Hydrogenation Catalysis. Organometallics 2021, 40, 2371–2374. [Google Scholar] [CrossRef]
- Tarannam, N.; Voelkel, M.H.; Huber, H.; Matile, S.; Kozuch, S. Chalcogen vs Halogen Bonding Catalysis in a Water-Bridge-Cocatalyzed Nitro-Michael Reaction. J. Org. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Benyu, Z.; Gabbai, F.P. Anion Chelation via Double Chalcogen Bonding: The Case of a Bis-telluronium Dication and Its Application in Electrophilic Catalysis via Metal-Chloride Bond Activation. J. Am. Chem. Soc. 2021, 143, 8625–8630. [Google Scholar]
- Xiangjin, K.; Pan-Pan, Z.; Yao, W. Chalcogen···π Bonding Catalysis. Angew. Chem. Int. Ed. 2021, 60, 9395–9400. [Google Scholar]
- Steinke, T.; Wonner, P.; Engelape, E.; Huber, H.; Matile, S. Catalytic Activation of a Carbon–Chloride Bond by Dicationic Tellurium-Based Chalcogen Bond Donors. Synthesis 2021, 53, 2043–2050. [Google Scholar]
- Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Chiral chalcogen bond donors based on the 4,4′-bipyridine scaffold. Molecules 2019, 24, 4484. [Google Scholar] [CrossRef] [Green Version]
- Wonner, P.; Steinke, T.; Vogel, L.; Huber, H.; Matile, S. Carbonyl Activation by Selenium- and Tellurium-Based Chalcogen Bonding in a Michael Addition Reaction. Chem. Eur. J. 2020, 26, 1258–1262. [Google Scholar] [CrossRef]
- Wonner, P.; Steinke, T.; Huber, H.; Matile, S. Activation of Quinolines by Cationic Chalcogen Bond Donors. Synlett 2019, 30, 1673–1678. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pn | α | RvdW | MEP (PnF3) |
|---|---|---|---|
| N | 5.3 | 1.55 | 15.9 |
| P | 16.9 | 1.80 | 27.4 |
| As | 21.6 | 1.85 | 38.5 |
| Sb | 30.8 | 2.06 | 46.7 |
| Pn-Catalyst | KD | ||||
| P(PFP)3 | N/A | ||||
| As(PFP)3 | 13,300 ± 800 | ||||
| Sb(Ph)3 | N/A | ||||
| Sb(Ph)2PFP | N/A | Ch-Catalyst | KD | Ha-Catalyst | KD |
| SbPh(PFP)2 | 570 ± 70 | Se(PFP)2 | 27,000 ± 4000 | BrPFP | N/A |
| Sb(PFP)3 | 19 ± 7 | Te(PFP)2 | 470 ± 70 | IPFP | 1370 ± 30 |
| Ch | α | RvdW | MEP (ChF2) |
|---|---|---|---|
| O | 3.0 | 1.52 | 16.8 |
| S | 11.8 | 1.80 | 35.6 |
| Se | 17.5 | 1.90 | 44.9 |
| Te | 25.9 | 2.06 | 52.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frontera, A.; Bauza, A. On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis. Int. J. Mol. Sci. 2021, 22, 12550. https://doi.org/10.3390/ijms222212550
Frontera A, Bauza A. On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis. International Journal of Molecular Sciences. 2021; 22(22):12550. https://doi.org/10.3390/ijms222212550
Chicago/Turabian StyleFrontera, Antonio, and Antonio Bauza. 2021. "On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis" International Journal of Molecular Sciences 22, no. 22: 12550. https://doi.org/10.3390/ijms222212550
APA StyleFrontera, A., & Bauza, A. (2021). On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis. International Journal of Molecular Sciences, 22(22), 12550. https://doi.org/10.3390/ijms222212550