
Priming, Triggering, Adaptation and Senescence (PTAS): A Hypothesis for a Common Damage Mechanism of Steatohepatitis



Abstract
:1. Introduction
1.1. What Is Steatohepatitis?
1.2. Current State of Knowledge about the Molecular Disease Mechanism
1.3. A Mechanistic Framework Is Urgently Needed
1.4. How Can a Mechanistic Framework Be Found?
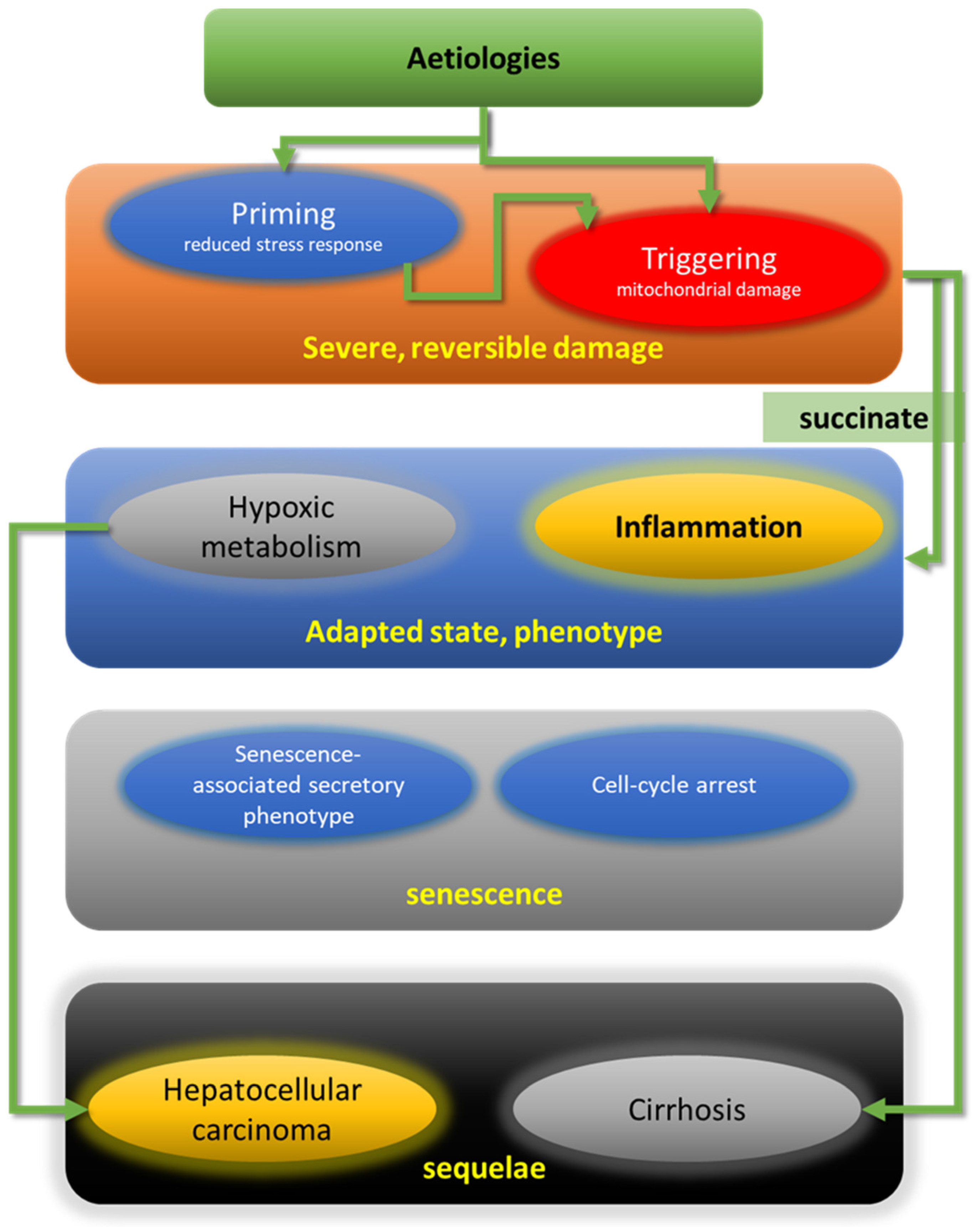
2. A Proposed Common Damage Mechanism (CDM) of SH: Priming/Sensitizing, Triggering, Adaptation and Senescence/Sequelae (PTAS)
3. Experimental Evidence Supporting PTAS
3.1. Priming Leads to Compromised Stress Defence

{kind=link}
| Factor | Mechanism | Model |
|---|---|---|
| Porphyrinogens (DDC, griseofulvin); liver toxins | AhR ↑ → PPARα ↓ & c-Myc ↓ → Nrf2-dependent genes ↓ | Intoxication mouse models [16,28,50,51,52,53,61,62]; DILI [12,13,14] |
| High-fat diet 1 | AhR ↑ → PPARα ↓ & c-Myc ↓ → Nrf2-dependent genes ↓ | HFD mouse model; human NASH [16,63,64,65,66,67,68] |
| High-fat diet 1 | Palmitoyl-CoA → NNT inhibition → NADPHmito ↓ → GSHmito | HFD mouse model; human NASH [69,70,71,72,73] |
| Excess keratin 8 | Impaired mitochondrial QC via Pirh2 | Keratin 18−/− mouse model, human NASH [16,59,74,75,76,77,78,79,80,81] |
3.1.1. Mechanistic Implications of PPARα Downregulation
3.1.2. Priming in Human SH
3.1.3. Priming Is a Factor Determining Sexual Dimorphism
3.2. Triggering Induces Severe Hepatocellular Injury
3.2.1. Triggering in Human SH
3.2.2. Triggering by Mitochondrial Dysfunction May Involve the Intermediate Filament Cytoskeleton
3.3. Adaptation—Mitigation of Acute Hepatocyte Damage and Involvement of Other Cell Types
Mitigation of Damage and Restoration of Stress Response
3.4. Senescence Prevents Neoplastic Transformation
3.5. Sequelae of SH—Cirrhosis and HCC
4. Discussion: Open Questions
4.1. Open Questions Regarding Priming and Triggering Events
4.2. Open Questions Regarding Adaptation
4.3. Open Questions Regarding Senescence
5. Conclusions: What We Presently Know and Do Not Know, and Some Diagnostic and Therapeutic Options
Diagnostic and Therapeutic Options
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geier, A.; Tiniakos, D.; Denk, H.; Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 2021, 70, 1570–1579. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M.; Neuschwander-Tetri, B.A.; Burt, A.D. Fatty liver disease: Alcoholic and non-alcoholic. In MacSween’s Pathology of the Liver, 6th ed.; Burt, A.D., Portmann, B.D.F.L., Eds.; Churchill Livingstone/Elsevier: London, UK, 2012; pp. 293–359. [Google Scholar]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E. Histopathology, grading and staging of nonalcoholic fatty liver disease. Minerva Gastroenterol. Dietol. 2017, 64, 28–38. [Google Scholar] [CrossRef]
- Nalbantoglu, I.L.; Brunt, E.M. Role of liver biopsy in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 9026–9037. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Working Group; Association of Pathologists; The Japan Society of Hepatology. Pathological Findings of NASH and NAFLD: For Guidebook of NASH and NAFLD, 2015: The Japan Society of Hepatology. Hepatol. Res. 2017, 47, 3–10. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Animal models of drug-induced liver injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1031–1039. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Biomarkers of drug-induced liver injury: Progress and utility in research, medicine, and regulation. Expert Rev. Mol. Diagn. 2018, 18, 797–807. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr. Pharmacol. Rep. 2018, 4, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Barve, S.; Kirpich, I.; Cave, M.C.; Marsano, L.S.; McClain, C.J. Alcoholic, Nonalcoholic, and Toxicant-Associated Steatohepatitis: Mechanistic Similarities and Differences. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denk, H.; Abuja, P.M.; Zatloukal, K. Animal models of NAFLD from the pathologist’s point of view. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [Green Version]
- Larter, C.Z.; Chitturi, S.; Heydet, D.; Farrell, G.C. A fresh look at NASH pathogenesis. Part 1: The metabolic movers. J. Gastroenterol. Hepatol. 2010, 25, 672–690. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Galloway, C.A.; Lee, H.; Brookes, P.S.; Yoon, Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G632–G641. [Google Scholar] [CrossRef] [Green Version]
- Perez-Carreras, M.; del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. 1), S20–S27. [Google Scholar] [CrossRef]
- Pessayre, D.; Berson, A.; Fromenty, B.; Mansouri, A. Mitochondria in steatohepatitis. Semin. Liver Dis. 2001, 21, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Nikam, A.; Patankar, J.V.; Lackner, C.; Schock, E.; Kratky, D.; Zatloukal, K.; Abuja, P.M. Transition between Acute and Chronic Hepatotoxicity in Mice Is Associated with Impaired Energy Metabolism and Induction of Mitochondrial Heme Oxygenase-1. PLoS ONE 2013, 8, e66094. [Google Scholar] [CrossRef] [Green Version]
- Singla, A.; Moons, D.S.; Snider, N.T.; Wagenmaker, E.R.; Jayasundera, V.B.; Omary, M.B. Oxidative stress, Nrf2 and keratin up-regulation associate with Mallory-Denk body formation in mouse erythropoietic protoporphyria. Hepatology 2012, 56, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Tirosh, O. Hypoxic Signaling and Cholesterol Lipotoxicity in Fatty Liver Disease Progression. Oxid. Med. Cell Longev. 2018, 2018, 2548154. [Google Scholar] [CrossRef] [PubMed]
- Polotsky, V.Y.; Patil, S.P.; Savransky, V.; Laffan, A.; Fonti, S.; Frame, L.A.; Steele, K.E.; Schweizter, M.A.; Clark, J.M.; Torbenson, M.S.; et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am. J. Respir. Crit. Care Med. 2009, 179, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, S.S.; Swiderska-Syn, M.; Sokol, R.J.; Halbower, A.C.; Capocelli, K.E.; Pan, Z.; Robbins, K.; Graham, B.; Diehl, A.M. Nocturnal Hypoxia Activation of the Hedgehog Signaling Pathway Affects Pediatric Nonalcoholic Fatty Liver Disease Severity. Hepatol. Commun. 2019, 3, 883–893. [Google Scholar] [CrossRef]
- Schafer, M.J.; Miller, J.D.; LeBrasseur, N.K. Cellular senescence: Implications for metabolic disease. Mol. Cell Endocrinol. 2017, 455, 93–102. [Google Scholar] [CrossRef]
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; le Couteur, D.G.; Cogger, V.C. Hallmarks of Aging in the Liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161. [Google Scholar] [CrossRef]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic senescence, the good and the bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef]
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef]
- Mills, E.L.; Harmon, C.; Jedrychowski, M.P.; Xiao, H.; Garrity, R.; Tran, N.V.; Bradshaw, G.A.; Fu, A.; Szpyt, J.; Reddy, A.; et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat. Metab. 2021, 3, 604–617. [Google Scholar] [CrossRef]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Kaser, S.; Tilg, H. Non-alcoholic steatohepatitis: A microbiota-driven disease. Trends Endocrinol. Metab. 2013, 24, 537–545. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Neuman, M.G.; French, S.W.; French, B.A.; Seitz, H.K.; Cohen, L.B.; Mueller, S.; Osna, N.A.; Kharbanda, K.K.; Seth, D.; Bautista, A.; et al. Alcoholic and non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2014, 97, 492–510. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Nikam, A.; Patankar, J.V.; Somlapura, M.; Lahiri, P.; Sachdev, V.; Kratky, D.; Denk, H.; Zatloukal, K.; Abuja, P.M. The PPARalpha Agonist Fenofibrate Prevents Formation of Protein Aggregates (Mallory-Denk bodies) in a Murine Model of Steatohepatitis-like Hepatotoxicity. Sci Rep. 2018, 8, 12964. [Google Scholar] [CrossRef] [PubMed]
- Stumptner, C.; Fuchsbichler, A.; Lehner, M.; Zatloukal, K.; Denk, H. Sequence of events in the assembly of Mallory body components in mouse liver: Clues to the pathogenesis and significance of Mallory body formation. J. Hepatol. 2001, 34, 665–675. [Google Scholar] [CrossRef]
- McCluskey, S.A.; Marks, G.S.; Sutherland, E.P.; Jacobsen, N.; Ortiz de Montellano, P.R. Ferrochelatase-inhibitory activity and N-alkylprotoporphyrin formation with analogues of 3,5-diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine (DDC) containing extended 4-alkyl groups: Implications for the active site of ferrochelatase. Mol. Pharmacol. 1986, 30, 352–357. [Google Scholar] [PubMed]
- McCluskey, S.A.; Marks, G.S.; Whitney, R.A.; Ortiz de Montellano, P.R. Differential inhibition of hepatic ferrochelatase by regioisomers of N-butyl-, N-pentyl-, N-hexyl-, and N-isobutylprotoporphyrin IX. Mol. Pharmacol. 1988, 34, 80–86. [Google Scholar]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell. 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Levy, S.; Forman, H.J. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life 2010, 62, 237–246. [Google Scholar] [CrossRef]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Du, Y.; Yuan, X.; Han, X.; Dong, Z.; Chen, X.; Wu, H.; Zhang, J.; Xu, L.; Han, C.; et al. Hepatic hypoxia-inducible factors inhibit PPARalpha expression to exacerbate acetaminophen induced oxidative stress and hepatotoxicity. Free Radic. Biol. Med. 2017, 110, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.L.; Li, L.Z.; Zhang, L.; Liu, Q.; Liu, K.; Li, P.; Liu, B.; Qi, L.W. Succinate accumulation impairs cardiac pyruvate dehydrogenase activity through GRP91-dependent and independent signaling pathways: Therapeutic effects of ginsenoside Rb1. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2835–2847. [Google Scholar] [CrossRef]
- Stumptner, C.; Fuchsbichler, A.; Zatloukal, K.; Denk, H. In vitro production of Mallory bodies and intracellular hyaline bodies: The central role of sequestosome 1/p62. Hepatology 2007, 46, 851–860. [Google Scholar] [CrossRef]
- Nakajima, T.; Kamijo, Y.; Tanaka, N.; Sugiyama, E.; Tanaka, E.; Kiyosawa, K.; Fukushima, Y.; Peters, J.M.; Gonzalez, F.J.; Aoyama, T. Peroxisome proliferator-activated receptor alpha protects against alcohol-induced liver damage. Hepatology 2004, 40, 972–980. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.G.; She, H.; Cheng, J.H.; French, S.W.; Koop, D.R.; Xiong, S.; Tsukamoto, H. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology 2005, 42, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Kucukoglu, O.; Guldiken, N.; Chen, Y.; Usachov, V.; El-Heliebi, A.; Haybaeck, J.; Denk, H.; Trautwein, C.; Strnad, P. High-fat diet triggers Mallory-Denk body formation through misfolding and crosslinking of excess keratin 8. Hepatology 2014, 60, 169–178. [Google Scholar] [CrossRef]
- Regnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouche, E.; Lasserre, F.; Naylies, C.; Betoulieres, C.; et al. Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef]
- Shaban, Z.; El-Shazly, S.; Abdelhady, S.; Fattouh, I.; Muzandu, K.; Ishizuka, M.; Kimura, K.; Kazusaka, A.; Fujita, S. Down regulation of hepatic PPARalpha function by AhR ligand. J. Vet. Med. Sci. 2004, 66, 1377–1386. [Google Scholar] [CrossRef] [Green Version]
- Sinal, C.J.; Bend, J.R. Aryl hydrocarbon receptor-dependent induction of cyp1a1 by bilirubin in mouse hepatoma hepa 1c1c7 cells. Mol. Pharmacol. 1997, 52, 590–599. [Google Scholar] [CrossRef]
- Bicego, R.; Francisco, A.; Ruas, J.S.; Siqueira-Santos, E.S.; Castilho, R.F. Undesirable effects of chemical inhibitors of NAD(P)(+) transhydrogenase on mitochondrial respiratory function. Arch. Biochem. Biophys. 2020, 692, 108535. [Google Scholar] [CrossRef]
- Gameiro, P.A.; Laviolette, L.A.; Kelleher, J.K.; Iliopoulos, O.; Stephanopoulos, G. Cofactor Balance by Nicotinamide Nucleotide Transhydrogenase (NNT) Coordinates Reductive Carboxylation and Glucose Catabolism in the Tricarboxylic Acid (TCA) Cycle. J. Biol. Chem. 2013, 288, 12967–12977. [Google Scholar] [CrossRef] [Green Version]
- McLennan, H.R.; Degli Esposti, M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr. 2000, 32, 153–162. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.F.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef]
- Duan, S.; Yao, Z.; Zhu, Y.; Wang, G.; Hou, D.; Wen, L.; Wu, M. The Pirh2-keratin 8/18 interaction modulates the cellular distribution of mitochondria and UV-induced apoptosis. Cell Death Differ. 2009, 16, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guldiken, N.; Zhou, Q.; Kucukoglu, O.; Rehm, M.; Levada, K.; Gross, A.; Kwan, R.; James, L.P.; Trautwein, C.; Omary, M.B.; et al. Human keratin 8 variants promote mouse acetaminophen hepatotoxicity coupled with c-jun amino-terminal kinase activation and protein adduct formation. Hepatology 2015, 62, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Hakem, A.; Bohgaki, M.; Lemmers, B.; Tai, E.; Salmena, L.; Matysiak-Zablocki, E.; Jung, Y.S.; Karaskova, J.; Kaustov, L.; Duan, S.; et al. Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genet. 2011, 7, e1002360. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.J.; Hakem, R.; Hakem, A. Pirh2: An E3 ligase with central roles in the regulation of cell cycle, DNA damage response, and differentiation. Cell Cycle 2013, 12, 2733–2737. [Google Scholar] [CrossRef] [Green Version]
- Kwan, R.; Hanada, S.; Harada, M.; Strnad, P.; Li, D.H.; Omary, M.B. Keratin 8 phosphorylation regulates its transamidation and hepatocyte Mallory-Denk body formation. FASEB J. 2012, 26, 2318–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magin, T.M.; Schroder, R.; Leitgeb, S.; Wanninger, F.; Zatloukal, K.; Grund, C.; Melton, D.W. Lessons from keratin 18 knockout mice: Formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J. Cell Biol. 1998, 140, 1441–1451. [Google Scholar] [CrossRef]
- Toivola, D.M.; Ku, N.O.; Resurreccion, E.Z.; Nelson, D.R.; Wright, T.L.; Omary, M.B. Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology 2004, 40, 459–466. [Google Scholar] [CrossRef]
- Zatloukal, K.; Stumptner, C.; Lehner, M.; Denk, H.; Baribault, H.; Eshkind, L.G.; Franke, W.W. Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am. J. Pathol. 2000, 156, 1263–1274. [Google Scholar] [CrossRef] [Green Version]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Bruick, R.K. Oxygen sensing in the hypoxic response pathway: Regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003, 17, 2614–2623. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Rodas, M.C.; Valenzuela, R.; Echeverria, F.; Rincon-Cervera, M.A.; Espinosa, A.; Illesca, P.; Munoz, P.; Corbari, A.; Romero, N.; Gonzalez-Manan, D.; et al. Supplementation with Docosahexaenoic Acid and Extra Virgin Olive Oil Prevents Liver Steatosis Induced by a High-Fat Diet in Mice through PPAR-alpha and Nrf2 Upregulation with Concomitant SREBP-1c and NF-kB Downregulation. Mol. Nutr. Food Res. 2017, 61, 1700479. [Google Scholar] [CrossRef]
- Maher, J.M.; Aleksunes, L.M.; Dieter, M.Z.; Tanaka, Y.; Peters, J.M.; Manautou, J.E.; Klaassen, C.D. Nrf2- and PPAR alpha-mediated regulation of hepatic Mrp transporters after exposure to perfluorooctanoic acid and perfluorodecanoic acid. Toxicol. Sci. 2008, 106, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Donepudi, A.C.; Moscovitz, J.E.; Slitt, A.L. Keap1-knockdown decreases fasting-induced fatty liver via altered lipid metabolism and decreased fatty acid mobilization from adipose tissue. PLoS ONE 2013, 8, e79841. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Klaassen, C.D. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARalpha-, and Nrf2-null mice. Drug Metab. Dispos. 2012, 40, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Brocker, C.N.; Takahashi, S.; Yagai, T.; Kim, T.; Xie, G.; Wang, H.; Qu, A.; Gonzalez, F.J. Keratin 23 Is a Peroxisome Proliferator-Activated Receptor Alpha-Dependent, MYC-Amplified Oncogene That Promotes Hepatocyte Proliferation. Hepatology 2019, 70, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Pagliarani, A. Nicotinamide Nucleotide Transhydrogenase as a Sensor of Mitochondrial Biology. Trends Cell Biol. 2020, 30, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.D.C.; Figueira, T.R.; Francisco, A.; Dal’Bo, G.A.; Ronchi, J.A.; Rovani, J.C.; Escanhoela, C.A.F.; Oliveira, H.C.F.; Castilho, R.F.; Vercesi, A.E. Redox imbalance due to the loss of mitochondrial NAD(P)-transhydrogenase markedly aggravates high fat diet-induced fatty liver disease in mice. Free Radic. Biol. Med. 2017, 113, 190–202. [Google Scholar] [CrossRef]
- Nakamura, A.; Terauchi, Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int. J. Mol. Sci. 2013, 14, 21240–21257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Publisher Correction: Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1755. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.H.; Chen, W.; Wang, L.L.; Sun, J.L.; Zhou, L.; Shi, Y.C.; Wang, C.H.; Zhong, B.H.; Shi, W.G.; Guo, Z.W. RLA8-A New and Highly Effective Quadruple PPAR-alpha/gamma/delta and GPR40 Agonist to Reverse Nonalcoholic Steatohepatitis and Fibrosis. J. Pharmacol. Exp. Ther. 2019, 369, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Nadeem, M.; Malik, M.I. Efficacy of elafibranor in patients with liver abnormalities especially non-alcoholic steatohepatitis: A systematic review and meta-analysis. Clin. J. Gastroenterol. 2021, 14, 1–8. [Google Scholar] [CrossRef]
- Smati, S.; Polizzi, A.; Fougerat, A.; Ellero-Simatos, S.; Blum, Y.; Lippi, Y.; Regnier, M.; Laroyenne, A.; Huillet, M.; Arif, M.; et al. Integrative study of diet-induced mouse models of NAFLD identifies PPARalpha as a sexually dimorphic drug target. Gut 2021. [Google Scholar] [CrossRef]
- Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Revelance to aging. J. Biol. Chem. 2001, 276, 48410–48416. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.H. Succinate as a Regulator of Hepatic Stellate Cells in Liver Fibrosis. Front. Endocrinol. 2018, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Grivennikova, V.G.; Kozlovsky, V.S.; Vinogradov, A.D. Respiratory complex II: ROS production and the kinetics of ubiquinone reduction. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 109–117. [Google Scholar] [CrossRef]
- Cunningham, C.C.; Coleman, W.B.; Spach, P.I. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol. 1990, 25, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Mezera, V.; Dighe, P.; Melov, S.; Gerencser, A.A.; Sweis, R.F.; Pliushchev, M.; Wang, Z.; Esbenshade, T.; McKibben, B.; et al. Superoxide produced by mitochondrial site IQ inactivates cardiac succinate dehydrogenase and induces hepatic steatosis in Sod2 knockout mice. Free Radic. Biol. Med. 2021, 164, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Correa, P.R.; Kruglov, E.A.; Thompson, M.; Leite, M.F.; Dranoff, J.A.; Nathanson, M.H. Succinate is a paracrine signal for liver damage. J. Hepatol. 2007, 47, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Le, C.T.; Sung, K.Y.; Choi, D.H.; Cho, E.H. Succinate induces hepatic fibrogenesis by promoting activation, proliferation, and migration, and inhibiting apoptosis of hepatic stellate cells. Biochem. Biophys. Res. Commun. 2018, 496, 673–678. [Google Scholar] [CrossRef]
- De Castro Fonseca, M.; Aguiar, C.J.; da Rocha Franco, J.A.; Gingold, R.N.; Leite, M.F. GPR91: Expanding the frontiers of Krebs cycle intermediates. Cell Commun. Signal. 2016, 14, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.H.; Woo, S.H.; Choi, D.H.; Cho, E.H. Succinate causes alpha-SMA production through GPR91 activation in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2015, 463, 853–858. [Google Scholar] [CrossRef]
- Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef]
- Macias-Ceja, D.C.; Ortiz-Masia, D.; Salvador, P.; Gisbert-Ferrandiz, L.; Hernandez, C.; Hausmann, M.; Rogler, G.; Esplugues, J.V.; Hinojosa, J.; Alos, R.; et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal. Immunol. 2019, 12, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.T.; Li, L.Z.; Yang, Y.L.; Yin, X.; Liu, Q.; Zhang, L.; Liu, K.; Liu, B.; Li, J.; Qi, L.W. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.K.; Imaizumi, N.; Chamberland, S.R.; Alder, N.N.; Boelsterli, U.A. Targeting mitochondria with methylene blue protects mice against acetaminophen-induced liver injury. Hepatology 2015, 61, 326–336. [Google Scholar] [CrossRef]
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine 2019, 49, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, A.; Li, X.; Jiang, Q. Irisin inhibition of growth hormone secretion in cultured tilapia pituitary cells. Mol. Cell Endocrinol. 2017, 439, 395–406. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Romano, A.D.; Giudetti, A.M.; Capitanio, N.; Petrella, A.; Vendemiale, G.; Altomare, E. Alterations of hepatic ATP homeostasis and respiratory chain during development of non-alcoholic steatohepatitis in a rodent model. Eur. J. Clin. Investig. 2008, 38, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Muller, M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Cong, W.N.; Tao, R.Y.; Tian, J.Y.; Liu, G.T.; Ye, F. The establishment of a novel non-alcoholic steatohepatitis model accompanied with obesity and insulin resistance in mice. Life Sci. 2008, 82, 983–990. [Google Scholar] [CrossRef]
- Berson, A.; de Beco, V.; Letteron, P.; Robin, M.A.; Moreau, C.; El Kahwaji, J.; Verthier, N.; Feldmann, G.; Fromenty, B.; Pessayre, D. Steatohepatitis-inducing drugs cause mitochondrial dysfunction and lipid peroxidation in rat hepatocytes. Gastroenterology 1998, 114, 764–774. [Google Scholar] [CrossRef]
- Denk, H.; Eckerstorfer, R. Colchicine-induced Mallory body formation in the mouse. Lab. Investig. 1977, 36, 563–565. [Google Scholar] [PubMed]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine—Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Riley, N.E.; Bardag-Gorce, F.; Montgomery, R.O.; Li, J.; Lungo, W.; Lue, Y.H.; French, S.W. Microtubules are required for cytokeratin aggresome (Mallory body) formation in hepatocytes: An in vitro study. Exp. Mol. Pathol. 2003, 74, 173–179. [Google Scholar] [CrossRef]
- Mahajan, V.; Klingstedt, T.; Simon, R.; Nilsson, K.P.; Thueringer, A.; Kashofer, K.; Haybaeck, J.; Denk, H.; Abuja, P.M.; Zatloukal, K. Cross beta-sheet conformation of keratin 8 is a specific feature of Mallory-Denk bodies compared with other hepatocyte inclusions. Gastroenterology 2011, 141, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Zatloukal, K.; Stumptner, C.; Kulaksiz, H.; Denk, H. Mallory-Denk-bodies: Lessons from keratin-containing hepatic inclusion bodies. Biochim. Biophys. Acta 2008, 1782, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Garcia, M.; Fusco, G.; de Simone, A. Membrane Interactions and Toxicity by Misfolded Protein Oligomers. Front. Cell Dev. Biol. 2021, 9, 642623. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, E.; Altman, B.J.; Seo, J.H.; Ghosh, J.C.; Kossenkov, A.V.; Tang, H.Y.; Krishn, S.R.; Languino, L.R.; Gabrilovich, D.I.; Speicher, D.W.; et al. Myc-mediated transcriptional regulation of the mitochondrial chaperone TRAP1 controls primary and metastatic tumor growth. J. Biol. Chem. 2019, 294, 10407–10414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarin, M.; Wang, Y.; Zhang, F.; Rothermund, K.; Zhang, Y.; Lu, J.; Sims-Lucas, S.; Beer-Stolz, D.; van Houten, B.E.; Vockley, J.; et al. Alterations in c-Myc phenotypes resulting from dynamin-related protein 1 (Drp1)-mediated mitochondrial fission. Cell Death Dis. 2013, 4, e670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Daks, A.; Petukhov, A.; Fedorova, O.; Shuvalov, O.; Kizenko, A.; Tananykina, E.; Vasileva, E.; Semenov, O.; Bottrill, A.; Barlev, N. The RNA-binding protein HuR is a novel target of Pirh2 E3 ubiquitin ligase. Cell Death Dis. 2021, 12, 581. [Google Scholar] [CrossRef]
- Ho, J.J.D.; Balukoff, N.C.; Theodoridis, P.R.; Wang, M.; Krieger, J.R.; Schatz, J.H.; Lee, S. A network of RNA-binding proteins controls translation efficiency to activate anaerobic metabolism. Nat. Commun. 2020, 11, 2677. [Google Scholar] [CrossRef]
- Marchesi, N.; Thongon, N.; Pascale, A.; Provenzani, A.; Koskela, A.; Korhonen, E.; Smedowski, A.; Govoni, S.; Kauppinen, A.; Kaarniranta, K.; et al. Autophagy Stimulus Promotes Early HuR Protein Activation and p62/SQSTM1 Protein Synthesis in ARPE-19 Cells by Triggering Erk1/2, p38(MAPK), and JNK Kinase Pathways. Oxid. Med. Cell Longev. 2018, 2018, 4956080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viiri, J.; Amadio, M.; Marchesi, N.; Hyttinen, J.M.; Kivinen, N.; Sironen, R.; Rilla, K.; Akhtar, S.; Provenzani, A.; D’Agostino, V.G.; et al. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS ONE 2013, 8, e69563. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Jiang, X.; Cui, X.; Wang, J.; Liu, S.; Li, H.; Yang, J.; Zhang, C.; Zhang, W. Smooth muscle-specific HuR knockout induces defective autophagy and atherosclerosis. Cell Death Dis. 2021, 12, 385. [Google Scholar] [CrossRef]
- Tian, M.; Wang, J.; Liu, S.; Li, X.; Li, J.; Yang, J.; Zhang, C.; Zhang, W. Hepatic HuR protects against the pathogenesis of non-alcoholic fatty liver disease by targeting PTEN. Cell Death Dis. 2021, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C. Succinate in ischemia: Where does it come from? Int. J. Biochem. Cell Biol. 2019, 115, 105580. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [Green Version]
- Matlac, D.M.; Hadrava Vanova, K.; Bechmann, N.; Richter, S.; Folberth, J.; Ghayee, H.K.; Ge, G.B.; Abunimer, L.; Wesley, R.; Aherrahrou, R.; et al. Succinate Mediates Tumorigenic Effects via Succinate Receptor 1: Potential for New Targeted Treatment Strategies in Succinate Dehydrogenase Deficient Paragangliomas. Front. Endocrinol. 2021, 12, 589451. [Google Scholar] [CrossRef]
- Hackenbeck, T.; Knaup, K.X.; Schietke, R.; Schodel, J.; Willam, C.; Wu, X.; Warnecke, C.; Eckardt, K.U.; Wiesener, M.S. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle 2009, 8, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.J.D.; Schatz, J.H.; Uniacke, J.; Lee, S. Jekyll and Hyde: Activating the Hypoxic Translational Machinery. Trends Biochem. Sci. 2021, 46, 171–174. [Google Scholar] [CrossRef]
- Ho, J.J.D.; Balukoff, N.C.; Cervantes, G.; Malcolm, P.D.; Krieger, J.R.; Lee, S. Oxygen-Sensitive Remodeling of Central Carbon Metabolism by Archaic eIF5B. Cell Rep. 2018, 22, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Elorza, A.; Soro-Arnaiz, I.; Melendez-Rodriguez, F.; Rodriguez-Vaello, V.; Marsboom, G.; de Carcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordonez, A.; Serrano-Oviedo, L.; et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwano, A.; Tanaka, M.; Suzuki, H.; Kurokawa, M.; Imoto, K.; Tashiro, S.; Goya, T.; Kohjima, M.; Kato, M.; Ogawa, Y. Upregulated expression of hypoxia reactive genes in peripheral blood mononuclear cells from chronic liver disease patients. Biochem. Biophys. Rep. 2021, 27, 101068. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, A.; Kurokawa, M.; Kohjima, M.; Imoto, K.; Tashiro, S.; Suzuki, H.; Tanaka, M.; Okada, S.; Kato, M.; Ogawa, Y. Microcirculatory disturbance in acute liver injury. Exp. Ther. Med. 2021, 21, 596. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Nan, L.; Wu, Y.; Bardag-Gorce, F.; Li, J.; French, B.A.; Fu, A.N.; Francis, T.; Vu, J.; French, S.W. p62 is involved in the mechanism of Mallory body formation. Exp. Mol. Pathol. 2004, 77, 168–175. [Google Scholar] [CrossRef]
- Lahiri, P.; Schmidt, V.; Smole, C.; Kufferath, I.; Denk, H.; Strnad, P.; Rulicke, T.; Frohlich, L.F.; Zatloukal, K. p62/Sequestosome-1 Is Indispensable for Maturation and Stabilization of Mallory-Denk Bodies. PLoS ONE 2016, 11, e0161083. [Google Scholar] [CrossRef]
- Manley, S.; Williams, J.A.; Ding, W.X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp. Biol. Med. 2013, 238, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Denk, H.; Stumptner, C.; Abuja, P.M.; Zatloukal, K. Sequestosome 1/p62-related pathways as therapeutic targets in hepatocellular carcinoma. Expert Opin. Ther. Targets 2019, 23, 393–406. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.X. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ni, H.M.; Chao, X.; Ma, X.; Rodriguez, Y.A.; Chavan, H.; Wang, S.; Krishnamurthy, P.; Dobrowsky, R.; Xu, D.X.; et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol. 2019, 22, 101148. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Lee, D.H.; Lee, Y.S.; Oh, E.; Bae, K.H.; Oh, K.J.; Kim, H.; Bae, S.H. Dual roles of ULK1 (unc-51 like autophagy activating kinase 1) in cytoprotection against lipotoxicity. Autophagy 2020, 16, 86–105. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Tang, Z.; Chen, D.; Moughon, D.; Ding, X.; Chen, S.; Zhu, M.; Zhong, Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010, 6, 614–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Itoh, K.; Yamamoto, M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002, 348, 182–190. [Google Scholar]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Kageyama, S.; Saito, T.; Obata, M.; Koide, R.H.; Ichimura, Y.; Komatsu, M. Negative regulation of the Keap1-Nrf2 pathway by a p62/Sqstm1 splicing variant. Mol. Cell Biol. 2018, 38, 00642-17. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Tabas, I.; Schwabe, R.F.; Pajvani, U.B. Maladaptive regeneration—The reawakening of developmental pathways in NASH and fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 131–142. [Google Scholar] [CrossRef]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdelho Machado, M.; Diehl, A.M. Role of Hedgehog Signaling Pathway in NASH. Int. J. Mol. Sci. 2016, 17, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Yan, Y.; Gius, D.R.; Vassilopoulos, A. Metabolic regulation of Sirtuins upon fasting and the implication for cancer. Curr. Opin. Oncol. 2013, 25, 630–636. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazi, E.; Zaouali, M.A.; Bejaoui, M.; Folch-Puy, E.; Ben Abdennebi, H.; Rosello-Catafau, J. Role of sirtuins in ischemia-reperfusion injury. World J. Gastroenterol. 2013, 19, 7594–7602. [Google Scholar] [CrossRef] [Green Version]
- Hubbi, M.E.; Hu, H.; Kshitiz; Gilkes, D.M.; Semenza, G.L. Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J. Biol. Chem. 2013, 288, 20768–20775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.; Pietrosi, G.; Hoare, M.; Jupp, J.; Marshall, A.; Verrill, C.; Davies, S.; Bateman, A.; Sheron, N.; Allison, M.; et al. Hepatocyte expression of the senescence marker p21 is linked to fibrosis and an adverse liver-related outcome in alcohol-related liver disease. PLoS ONE 2013, 8, e72904. [Google Scholar] [CrossRef] [Green Version]
- Aravinthan, A.; Scarpini, C.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol. 2013, 58, 549–556. [Google Scholar] [CrossRef]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Jurk, D. Senescence explains age- and obesity-related liver steatosis. Cell Stress 2017, 1, 70–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Cazzagon, N.; Sarcognato, S.; Floreani, A.; Corra, G.; de Martin, S.; Guzzardo, V.; Russo, F.P.; Guido, M. Cholangiocyte senescence in primary sclerosing cholangitis is associated with disease severity and prognosis. JHEP Rep. 2021, 3, 100286. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Gonzalez, S.; Lu, W.Y.; Raven, A.; Dwyer, B.; Man, T.Y.; O’Duibhir, E.; Lewis, P.J.S.; Campana, L.; Kendall, T.J.; Bird, T.G.; et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 2018, 9, 1020. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Yi, W.; Lan, H.; Wen, Y.; Wang, Y.; He, D.; Bai, Z.; Zhang, Y.; Jiang, W.; Liu, B.; Shen, J.; et al. HO-1 overexpression alleviates senescence by inducing autophagy via the mitochondrial route in human nucleus pulposus cells. J. Cell Physiol. 2020, 235, 8402–8415. [Google Scholar] [CrossRef]
- Dou, F.; Wu, B.; Chen, J.; Liu, T.; Yu, Z.; Chen, C. PPARalpha Targeting GDF11 Inhibits Vascular Endothelial Cell Senescence in an Atherosclerosis Model. Oxid. Med. Cell Longev. 2021, 2021, 2045259. [Google Scholar] [CrossRef]
- Hanada, S.; Harada, M.; Abe, M.; Akiba, J.; Sakata, M.; Kwan, R.; Taniguchi, E.; Kawaguchi, T.; Koga, H.; Nagata, E.; et al. Aging modulates susceptibility to mouse liver Mallory-Denk body formation. J. Histochem. Cytochem. 2012, 60, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Li, H.Y.; Zhang, L.; Zhou, Y.; Wu, J. Hedgehog Signaling, a Critical Pathway Governing the Development and Progression of Hepatocellular Carcinoma. Cells 2021, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.S.; Guy, C.D.; Chen, Y.; Lu, J.; Swiderska-Syn, M.; Michelotti, G.A.; Karaca, G.; Xie, G.; Kruger, L.; Syn, W.K.; et al. Paracrine Hedgehog signaling drives metabolic changes in hepatocellular carcinoma. Cancer Res. 2012, 72, 6344–6350. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, F.V.; Tardelli, M.; Herac, M.; Claudel, T.; Trauner, M. Metabolic regulation of hepatic PNPLA3 expression and severity of liver fibrosis in patients with NASH. Liver Int. 2020, 40, 1098–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruschi, F.V.; Tardelli, M.; Einwallner, E.; Claudel, T.; Trauner, M. PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells. Int. J. Mol. Sci. 2020, 21, 8711. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Reddy, J.K. PPARalpha in the pathogenesis of fatty liver disease. Hepatology 2004, 40, 783–786. [Google Scholar] [CrossRef]
- Souza-Mello, V. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 1012–1019. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; McIntosh, A.L.; Martin, G.G.; Petrescu, A.D.; Landrock, K.K.; Landrock, D.; Kier, A.B.; Schroeder, F. Inhibitors of Fatty Acid Synthesis Induce PPAR alpha-Regulated Fatty Acid beta*Oxidative Genes: Synergistic Roles of L-FABP and Glucose. PPAR Res. 2013, 2013, 865604. [Google Scholar] [CrossRef] [Green Version]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef] [Green Version]
- Mossa, A.H.; Velasquez Flores, M.; Cammisotto, P.G.; Campeau, L. Succinate, increased in metabolic syndrome, activates GPR91 receptor signaling in urothelial cells. Cell Signal. 2017, 37, 31–39. [Google Scholar] [CrossRef]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens 2007, 20, 1209–1215. [Google Scholar] [CrossRef]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef]
- Sundaram, S.S.; Halbower, A.; Pan, Z.; Robbins, K.; Capocelli, K.E.; Klawitter, J.; Shearn, C.T.; Sokol, R.J. Nocturnal hypoxia-induced oxidative stress promotes progression of pediatric non-alcoholic fatty liver disease. J. Hepatol. 2016, 65, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, J.; Fu, H.; Li, Y.; Wang, L.; Luo, S.; Lu, H. Hypoxia exacerbates nonalcoholic fatty liver disease via the HIF-2alpha/PPARalpha pathway. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E710–E722. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.; Huang, J.; Li, Z.; Gong, Y.; Zou, B.; Liu, X.; Ding, L.; Li, P.; Zhu, Z.; et al. HIF-2alpha upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway. Aging 2019, 11, 10839–10860. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [Green Version]
- Ham, P.B., 3rd; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Wittig, I.; Heide, H.; Dehne, N.; Brune, B. Chronic hypoxia alters mitochondrial composition in human macrophages. Biochim. Biophys. Acta 2013, 1834, 2750–2760. [Google Scholar] [CrossRef]
- Keiran, N.; Ceperuelo-Mallafre, V.; Calvo, E.; Hernandez-Alvarez, M.I.; Ejarque, M.; Nunez-Roa, C.; Horrillo, D.; Maymo-Masip, E.; Rodriguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592. [Google Scholar] [CrossRef]
- Ferreira-Gonzalez, S.; Rodrigo-Torres, D.; Gadd, V.L.; Forbes, S.J. Cellular Senescence in Liver Disease and Regeneration. Semin. Liver Dis. 2021, 41, 50–66. [Google Scholar] [CrossRef]
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. 2021, 3, 100301. [Google Scholar] [CrossRef]
- Moustakas, I.I.; Katsarou, A.; Legaki, A.I.; Pyrina, I.; Ntostoglou, K.; Papatheodoridi, A.M.; Gercken, B.; Pateras, I.S.; Gorgoulis, V.G.; Koutsilieris, M.; et al. Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity. Int. J. Mol. Sci. 2021, 22, 3446. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Sanguino, E.; Roglans, N.; Alegret, M.; Sanchez, R.M.; Vazquez-Carrera, M.; Laguna, J.C. Atorvastatin reverses age-related reduction in rat hepatic PPARalpha and HNF-4. Br. J. Pharmacol. 2005, 145, 853–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Koteish, A.; Lin, H.; Huang, J.; Roskams, T.; Dawson, V.; Diehl, A.M. Oval cells compensate for damage and replicative senescence of mature hepatocytes in mice with fatty liver disease. Hepatology 2004, 39, 403–411. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Koch, A.; Seidler, S.; Trautwein, C.; Tacke, F. Circulating soluble urokinase plasminogen activator is elevated in patients with chronic liver disease, discriminates stage and aetiology of cirrhosis and predicts prognosis. Liver Int. 2012, 32, 500–509. [Google Scholar] [CrossRef]
- Wyss, R.K.; Mendez Carmona, N.; Arnold, M.; Segiser, A.; Mueller, M.; Dutkowski, P.; Carrel, T.P.; Longnus, S.L. Hypothermic, oxygenated perfusion (HOPE) provides cardioprotection via succinate oxidation prior to normothermic perfusion in a rat model of donation after circulatory death (DCD). Am. J. Transplant. 2021, 21, 1003–1011. [Google Scholar] [CrossRef]
- Fafian-Labora, J.A.; Rodriguez-Navarro, J.A.; O’Loghlen, A. Small Extracellular Vesicles Have GST Activity and Ameliorate Senescence-Related Tissue Damage. Cell Metab. 2020, 32, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Fafian-Labora, J.A.; O’Loghlen, A. Classical and Nonclassical Intercellular Communication in Senescence and Ageing. Trends Cell Biol. 2020, 30, 628–639. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Cocheme, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, B.D.; Herlein, J.A.; Guo, D.F.; Kulkarni, C.; Weidemann, B.J.; Yu, L.; Grobe, J.L.; Rahmouni, K.; Kerns, R.J.; Sivitz, W.I. A mitochondrial-targeted coenzyme q analog prevents weight gain and ameliorates hepatic dysfunction in high-fat-fed mice. J. Pharmacol. Exp. Ther. 2014, 351, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, B.D.; Herlein, J.A.; Yorek, M.A.; Fenner, A.M.; Kerns, R.J.; Sivitz, W.I. Bioenergetic effects of mitochondrial-targeted coenzyme Q analogs in endothelial cells. J. Pharmacol. Exp. Ther. 2012, 342, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2015, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Sheard, M.A.; Vojtesek, B.; Simickova, M.; Valik, D. Release of cytokeratin-18 and -19 fragments (TPS and CYFRA 21-1) into the extracellular space during apoptosis. J. Cell Biochem. 2002, 85, 670–677. [Google Scholar] [CrossRef]
- Tarantino, G.; Conca, P.; Coppola, A.; Vecchione, R.; di Minno, G. Serum concentrations of the tissue polypeptide specific antigen in patients suffering from non-alcoholic steatohepatitis. Eur. J. Clin. Investig. 2007, 37, 48–53. [Google Scholar] [CrossRef]
| Factor | Mechanism | Model |
|---|---|---|
| Porphyrinogens (DDC, griseofulvin); liver toxins | Inhibition of ferrochelatase (haem deficiency) | Intoxication mouse models [28,50,51,52,53,100]; DILI [12,13,14] |
| Excess keratin 8 1 | Impaired mitochondrial QC via Pirh2 | Keratin 18−/− mouse model, HFD mouse model, human NASH [65,74,78,79,80,81] |
| HFD 2 | Increased ROS production by β-oxidation of fatty acids | HFD model, human NASH [18,22,27,31,93,117] |
| Stage | Outcome | Effect |
|---|---|---|
| priming | Persistently reduced PPARα | pseudohypoxia |
| triggering | Damage to mitochondrial ETC Reduced mitochondrial QC succinate ↑ | DAMP, ROS ↑, succinate ↑, pseudohypoxia, cell cycle arrest DAMP, stellate cell activation, immune response, inflammation |
| Keratin 8 excess | Reduced mitochondrial QC, cell cycle arrest 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuja, P.M.; Zatloukal, K.; Denk, H. Priming, Triggering, Adaptation and Senescence (PTAS): A Hypothesis for a Common Damage Mechanism of Steatohepatitis. Int. J. Mol. Sci. 2021, 22, 12545. https://doi.org/10.3390/ijms222212545
Abuja PM, Zatloukal K, Denk H. Priming, Triggering, Adaptation and Senescence (PTAS): A Hypothesis for a Common Damage Mechanism of Steatohepatitis. International Journal of Molecular Sciences. 2021; 22(22):12545. https://doi.org/10.3390/ijms222212545
Chicago/Turabian StyleAbuja, Peter M., Kurt Zatloukal, and Helmut Denk. 2021. "Priming, Triggering, Adaptation and Senescence (PTAS): A Hypothesis for a Common Damage Mechanism of Steatohepatitis" International Journal of Molecular Sciences 22, no. 22: 12545. https://doi.org/10.3390/ijms222212545
APA StyleAbuja, P. M., Zatloukal, K., & Denk, H. (2021). Priming, Triggering, Adaptation and Senescence (PTAS): A Hypothesis for a Common Damage Mechanism of Steatohepatitis. International Journal of Molecular Sciences, 22(22), 12545. https://doi.org/10.3390/ijms222212545