
Enhancer RNA Profiling in Smoking and HPV Associated HNSCC Reveals Associations to Key Oncogenes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
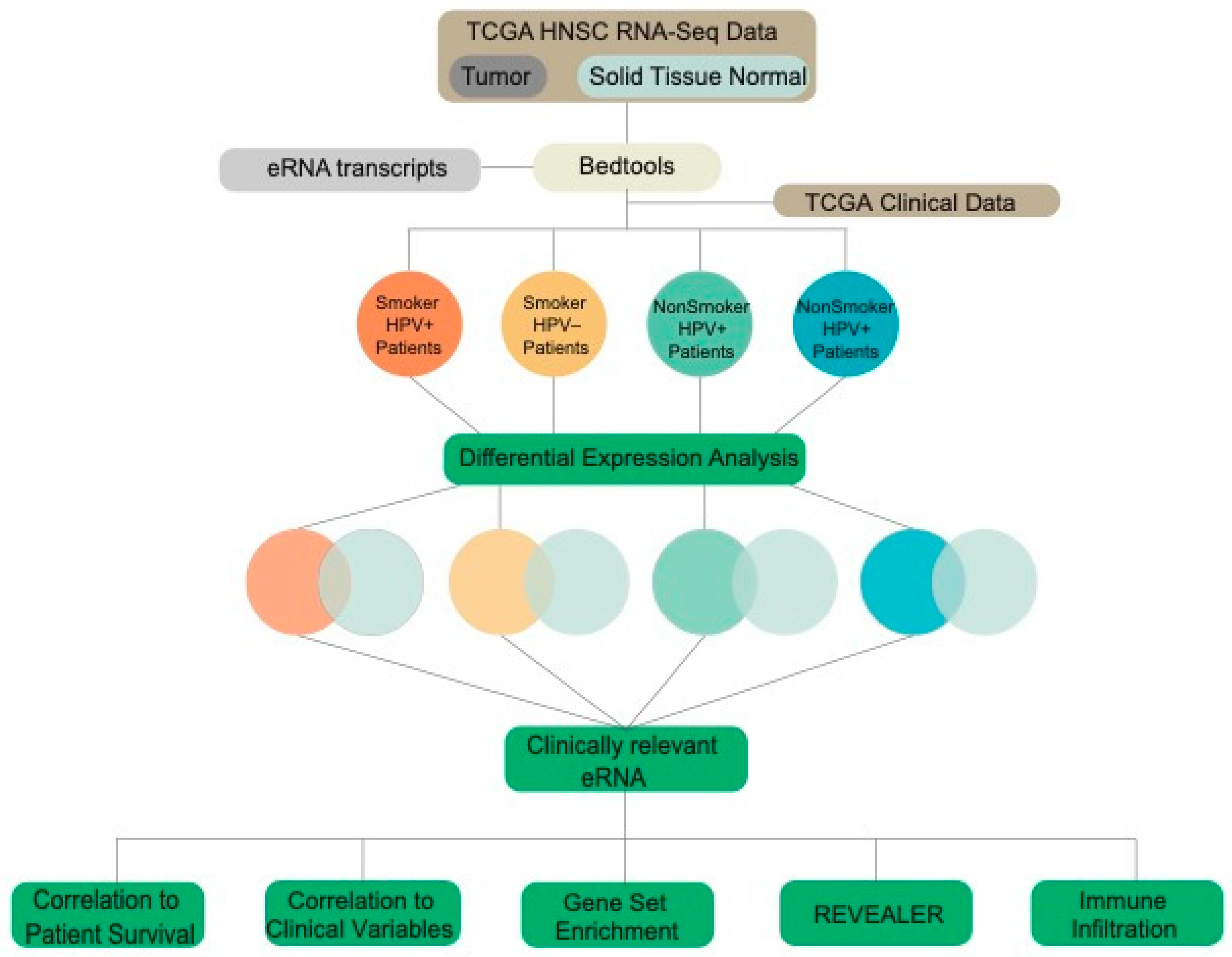
2.1. TCGA Data Acquisition and Extraction of eRNA Reads
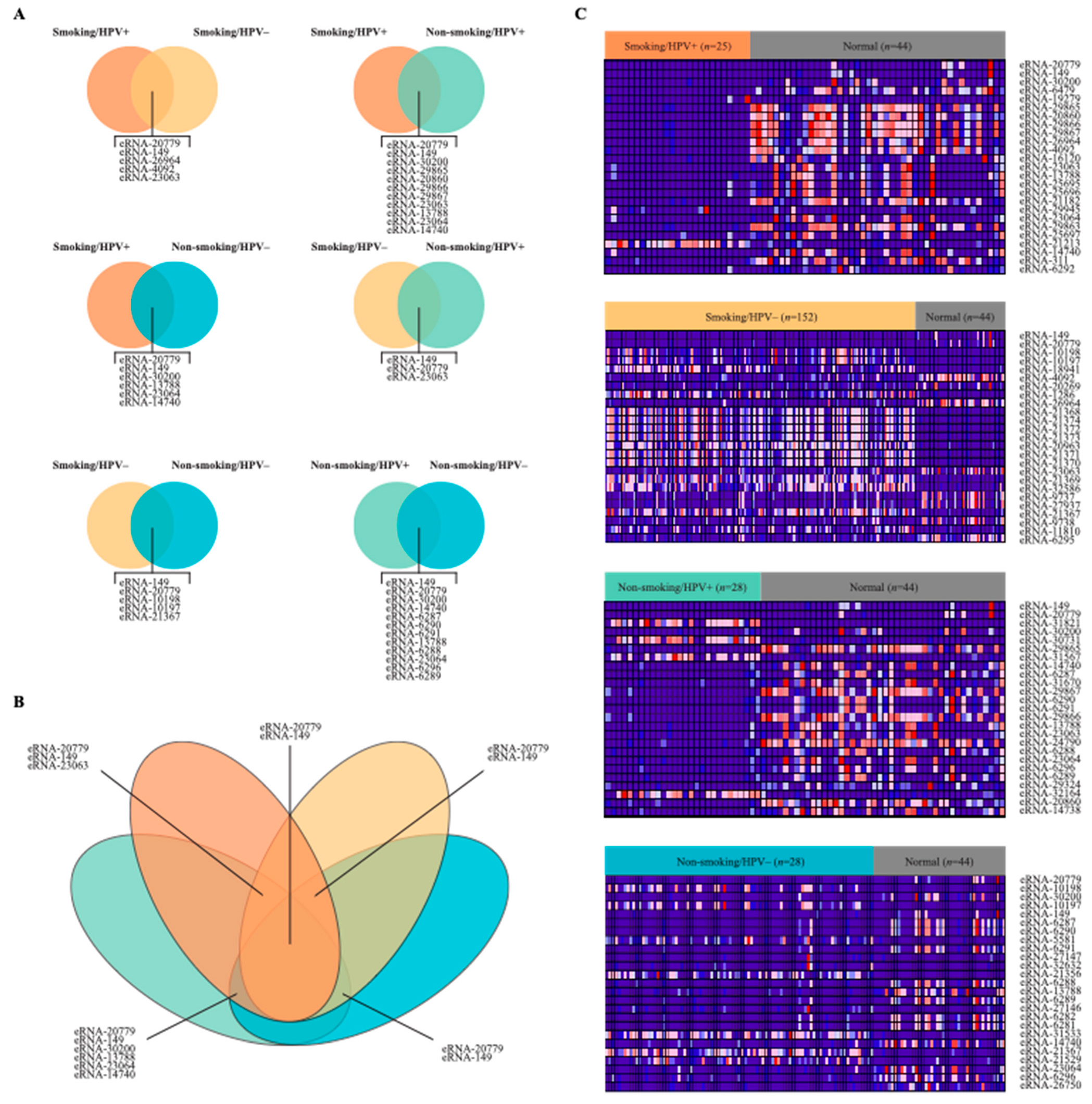
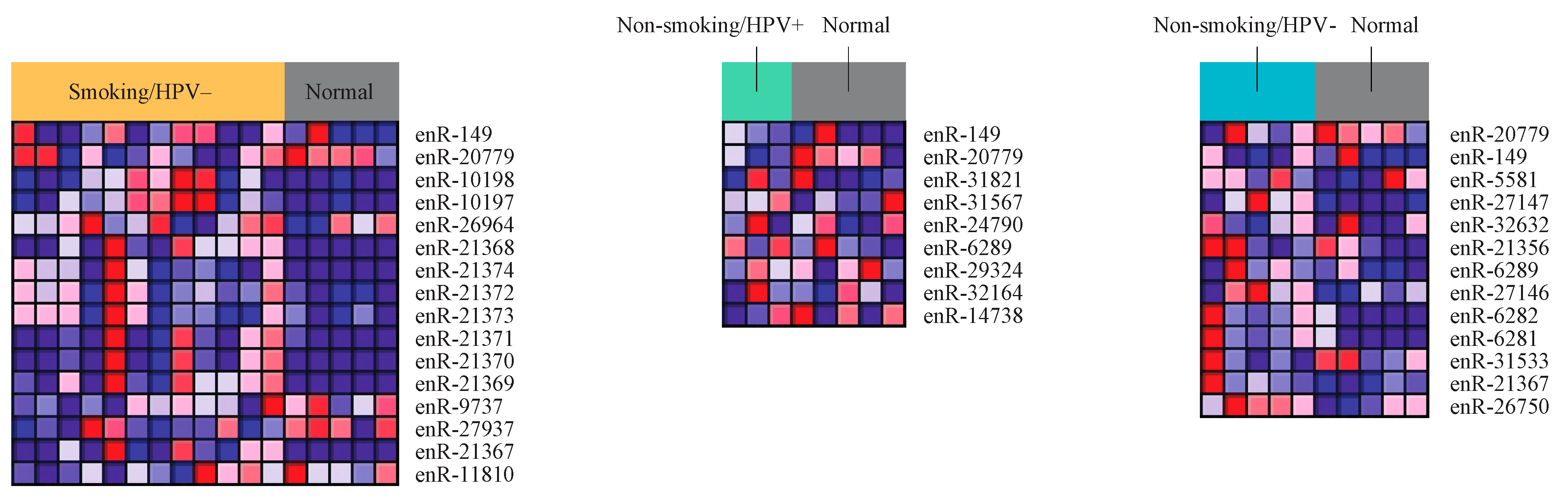
2.2. Differential Expression of eRNA between Cancer and Normal Tissue Samples
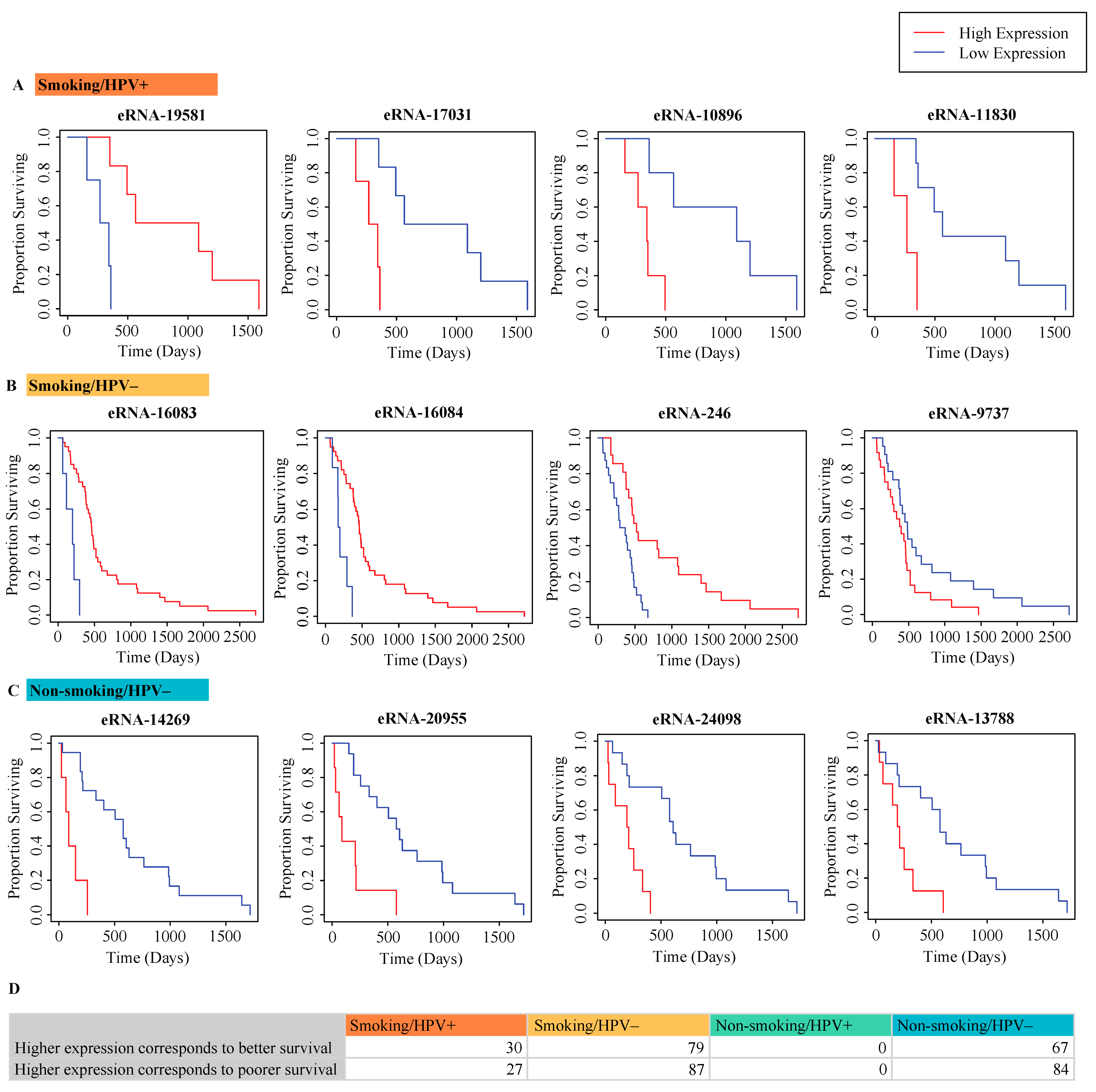
2.3. Correlation of eRNA to Survival of Patients
2.4. Correlation of eRNA to Clinical Variables
2.5. Correlation of eRNA to Immune Infiltration
2.6. Correlation of eRNA to Cancer and Immune-Associated Signatures
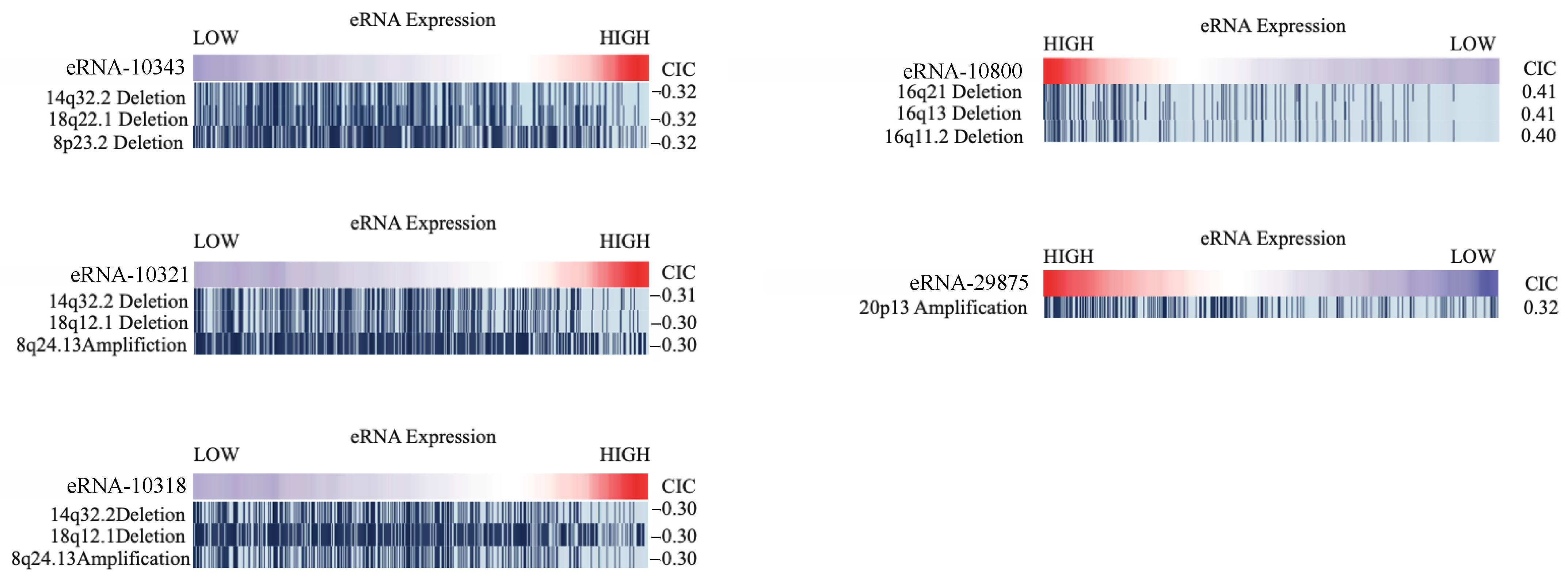
2.7. Correlation of eRNA to Genomic Alterations
2.8. Differential Expression of eRNA between Cancer and Normal Samples from Secondary Validation Dataset
3. Discussion
4. Materials and Methods
4.1. Data Acquisition from TCGA
4.2. Extraction of eRNA Reads
4.3. Differential Expression of eRNA between Cancer and Normal Tissue Samples
4.4. Correlation of eRNA to Survival of Patients
4.5. Correlation of eRNA to Clinical Variables
4.6. Correlation of eRNA to Immune Infiltration
4.7. Correlation of eRNA to Cancer and Immune-Associated Signatures
4.8. Correlation of eRNA to Genomic Alterations
4.9. Validation of TCGA Results with Secondary Dataset
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Sharma, A.; Schmitt, N.C.; Johnson, J.T.; Ferris, R.L.; Duvvuri, U.; Kim, S. A 20-Year Review of 75 Cases of Salivary Duct Carcinoma. JAMA Otolaryngol. Head Neck Surg. 2016, 142, 489–495. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Cancer Stat Facts: Oral Cavity and Pharynx Cancer. [Surveillance, Epidemiology, and End Results Program]. Available online: https://seer.cancer.gov/statfacts/html/oralcav.html (accessed on 1 September 2021).
- Arnold, P.R.; Wells, A.D.; Li, X.C. Diversity and Emerging Roles of Enhancer RNA in Regulation of Gene Expression and Cell Fate. Front. Cell Dev. Biol. 2019, 7, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Zorca, C.E.; Traboulsi, T.; Noutahi, E.; Krause, M.R.; Mader, S.; Zenklusen, D. Single-cell profiling reveals that eRNA accumulation at enhancer-promoter loops is not required to sustain transcription. Nucleic Acids Res. 2017, 45, 3017–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azofeifa, J.G.; Allen, M.A.; Hendrix, J.R.; Read, T.; Rubin, J.D.; Dowell, R.D. Enhancer RNA profiling predicts transcription factor activity. Genome Res. 2018; 28, 334–344. [Google Scholar]
- Zhang, Z.; Lee, J.H.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 4562. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Tang, Q. Current Advances on the Important Roles of Enhancer RNAs in Molecular Pathways of Cancer. Int. J. Mol. Sci. 2021, 22, 5640. [Google Scholar] [CrossRef]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Blot, W.J.; McLaughlin, J.K.; Winn, D.M.; Austin, D.F.; Greenberg, R.S.; Preston-Martin, S.; Bernstein, L.; Schoenberg, J.B.; Stemhagen, A.; Fraumeni, J.F. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988, 48, 3282–3287. [Google Scholar]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Wyss, A.; Hashibe, M.; Chuang, S.C.; Lee, Y.C.; Zhang, Z.F.; Yu, G.P.; Winn, D.M.; Wei, Q.; Talamini, R.; Szeszenia-Dabrowska, N.; et al. Cigarette, cigar, and pipe smoking and the risk of head and neck cancers: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Am. J. Epidemiol. 2013, 178, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Lacko, M.; Braakhuis, B.J.; Sturgis, E.M.; Boedeker, C.C.; Suárez, C.; Rinaldo, A.; Ferlito, A.; Takes, R.P. Genetic susceptibility to head and neck squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, A.R.; Khariwala, S.S. Tobacco-related carcinogenesis in head and neck cancer. Cancer Metastasis Rev. 2017, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Morris, L.G.; Lee, W.; Chan, T.A. Unraveling the molecular genetics of head and neck cancer through genome-wide approaches. Genes Dis. 2014, 1, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Božinović, K.; Sabol, I.; Dediol, E.; Milutin Gašperov, N.; Manojlović, S.; Vojtechova, Z.; Tachezy, R.; Grce, M. Genome-wide miRNA profiling reinforces the importance of miR-9 in human papillomavirus associated oral and oropharyngeal head and neck cancer. Sci. Rep. 2019, 9, 2306. [Google Scholar] [CrossRef]
- Zou, A.E.; Ku, J.; Honda, T.K.; Yu, V.; Kuo, S.Z.; Zheng, H.; Xuan, Y.; Saad, M.A.; Hinton, A.; Brumund, K.T.; et al. Transcriptome sequencing uncovers novel long noncoding and small nucleolar RNAs dysregulated in head and neck squamous cell carcinoma. RNA 2015, 21, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Ienasescu, H.; Li, K.; Andersson, R.; Vitezic, M.; Rennie, S.; Chen, Y.; Vitting-Seerup, K.; Lagoni, E.; Boyd, M.; Bornholdt, J.; et al. On-the-fly selection of cell-specific enhancers, genes, miRNAs and proteins across the human body using SlideBase. Database 2016, 2016:baw144. [Google Scholar] [CrossRef] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Cillo, A.R.; Kürten, C.H.L.; Tabib, T.; Qi, Z.; Onkar, S.; Wang, T.; Liu, A.; Duvvuri, U.; Kim, S.; Soose, R.J.; et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199. [Google Scholar] [CrossRef]
- Ruffin, A.T.; Cillo, A.R.; Tabib, T.; Liu, A.; Onkar, S.; Kunning, S.R.; Lampenfeld, C.; Atiya, H.I.; Abecassis, I.; Kürten, C.H.L.; et al. B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat. Commun. 2021, 12, 3349. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Locally advanced squamous carcinoma of the head and neck. In 2014 Review of Cancer Medicines on the WHO List of Essential Medicines; Union for International Cancer Control: Geneva, Switzerland, 2014; pp. 1–8.
- Dhull, A.K.; Atri, R.; Dhankhar, R.; Chauhan, A.K.; Kaushal, V. Major Risk Factors in Head and Neck Cancer: A Retrospective Analysis of 12-Year Experiences. World J. Oncol. 2018, 9, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beachler, D.C.; Abraham, A.G.; Silverberg, M.J.; Jing, Y.; Fakhry, C.; Gill, M.J.; Dubrow, R.; Kitahata, M.M.; Klein, M.B.; Burchell, A.N.; et al. Incidence and risk factors of HPV-related and HPV-unrelated Head and Neck Squamous Cell Carcinoma in HIV-infected individuals. Oral Oncol. 2014, 50, 1169–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.P.; den Breems, N.Y.; Richa, T.; Nwagu, U.; Zhan, T.; Poojan, S.; Martinez-Outschoorn, U.; Johnson, J.M.; Luginbuhl, A.J.; Curry, J.M. Mutation signature analysis identifies increased mutation caused by tobacco smoke associated DNA adducts in larynx squamous cell carcinoma compared with oral cavity and oropharynx. Sci. Rep. 2019, 9, 19256. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, F.; Prokopec, S.D.; MacNeil, D.; Mundi, N.; Gameiro, S.F.; Howlett, C.; Stecho, W.; Plantinga, P.; Pinto, N.; Ruicci, K.M.; et al. Mutational analysis of head and neck squamous cell carcinoma stratified by smoking status. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Irimie, A.I.; Braicu, C.; Cojocneanu, R.; Magdo, L.; Onaciu, A.; Ciocan, C.; Mehterov, N.; Dudea, D.; Buduru, S.; Berindan-Neagoe, I. Differential Effect of Smoking on Gene Expression in Head and Neck Cancer Patients. Int. J. Environ. Res. Public Health 2018, 15. [Google Scholar] [CrossRef] [Green Version]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-Mediated Cytosine Deamination Links PIK3CA Helical Domain Mutations to Human Papillomavirus-Driven Tumor Development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, A.C.; Palma, D.A.; Chow, W.; Tan, S.; Rajakumar, C.; Rizzo, G.; Fung, K.; Kwan, K.; Wehrli, B.; Winquist, E.; et al. High frequency of activating PIK3CA mutations in human papillomavirus-positive oropharyngeal cancer. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Hajek, M.; Sewell, A.; Kaech, S.; Burtness, B.; Yarbrough, W.G.; Issaeva, N. TRAF3/CYLD mutations identify a distinct subset of human papillomavirus-associated head and neck squamous cell carcinoma. Cancer 2017, 123, 1778–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, D.N.; Van Waes, C.; Seiwert, T.Y. Genetic Landscape of Human Papillomavirus-Associated Head and Neck Cancer and Comparison to Tobacco-Related Tumors. J. Clin. Oncol. 2015, 33, 3227–3234. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Choi, B.Y.; Lee, M.H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Izreig, S.; Yarbrough, W.G.; Issaeva, N. NSD1 mutations by HPV status in head and neck cancer: Differences in survival and response to DNA-damaging agents. Cancers Head Neck 2019, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Botvinnik, O.B.; Abudayyeh, O.; Birger, C.; Rosenbluh, J.; Shrestha, Y.; Abazeed, M.E.; Hammerman, P.S.; DiCara, D.; Konieczkowski, D.J.; et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat. Biotechnol. 2016, 34, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Ha, B.; Greenbaum, J.A.; Shmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Altay, G.; McVicker, G.; et al. Database of Immune Cell EQTLs, Expression, Epigenomics. J. Immunol. 2019, 202 (Suppl. 1), 131.118. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shende, N.; Xu, J.; Li, W.T.; Liu, J.; Chakladar, J.; Brumund, K.T.; Ongkeko, W.M. Enhancer RNA Profiling in Smoking and HPV Associated HNSCC Reveals Associations to Key Oncogenes. Int. J. Mol. Sci. 2021, 22, 12546. https://doi.org/10.3390/ijms222212546
Shende N, Xu J, Li WT, Liu J, Chakladar J, Brumund KT, Ongkeko WM. Enhancer RNA Profiling in Smoking and HPV Associated HNSCC Reveals Associations to Key Oncogenes. International Journal of Molecular Sciences. 2021; 22(22):12546. https://doi.org/10.3390/ijms222212546
Chicago/Turabian StyleShende, Neil, Jingyue Xu, Wei Tse Li, Jeffrey Liu, Jaideep Chakladar, Kevin T. Brumund, and Weg M. Ongkeko. 2021. "Enhancer RNA Profiling in Smoking and HPV Associated HNSCC Reveals Associations to Key Oncogenes" International Journal of Molecular Sciences 22, no. 22: 12546. https://doi.org/10.3390/ijms222212546
APA StyleShende, N., Xu, J., Li, W. T., Liu, J., Chakladar, J., Brumund, K. T., & Ongkeko, W. M. (2021). Enhancer RNA Profiling in Smoking and HPV Associated HNSCC Reveals Associations to Key Oncogenes. International Journal of Molecular Sciences, 22(22), 12546. https://doi.org/10.3390/ijms222212546