
Protein Misfolding and Aggregation: The Relatedness between Parkinson’s Disease and Hepatic Endoplasmic Reticulum Storage Disorders


 ,
, 
Abstract
:1. Introduction
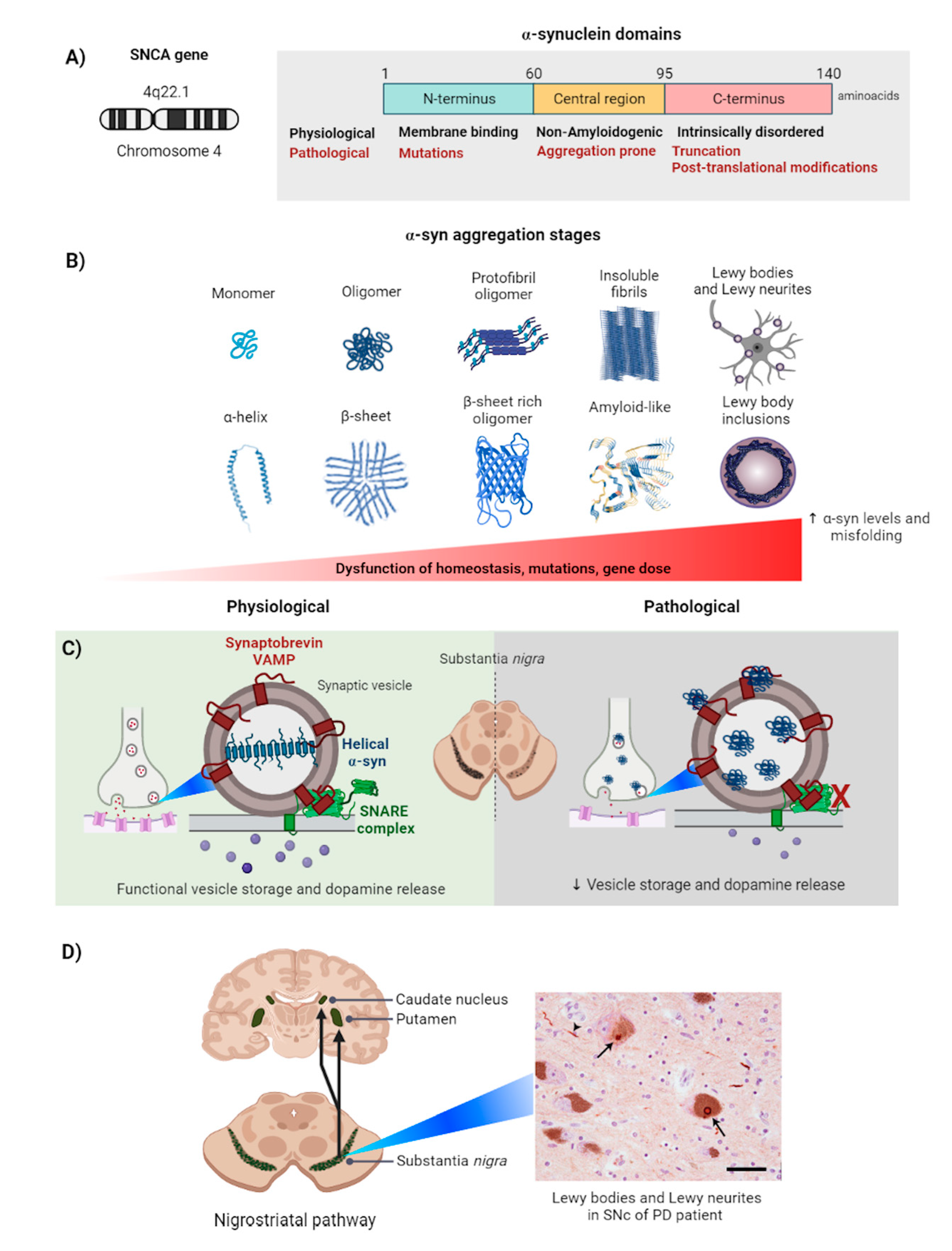
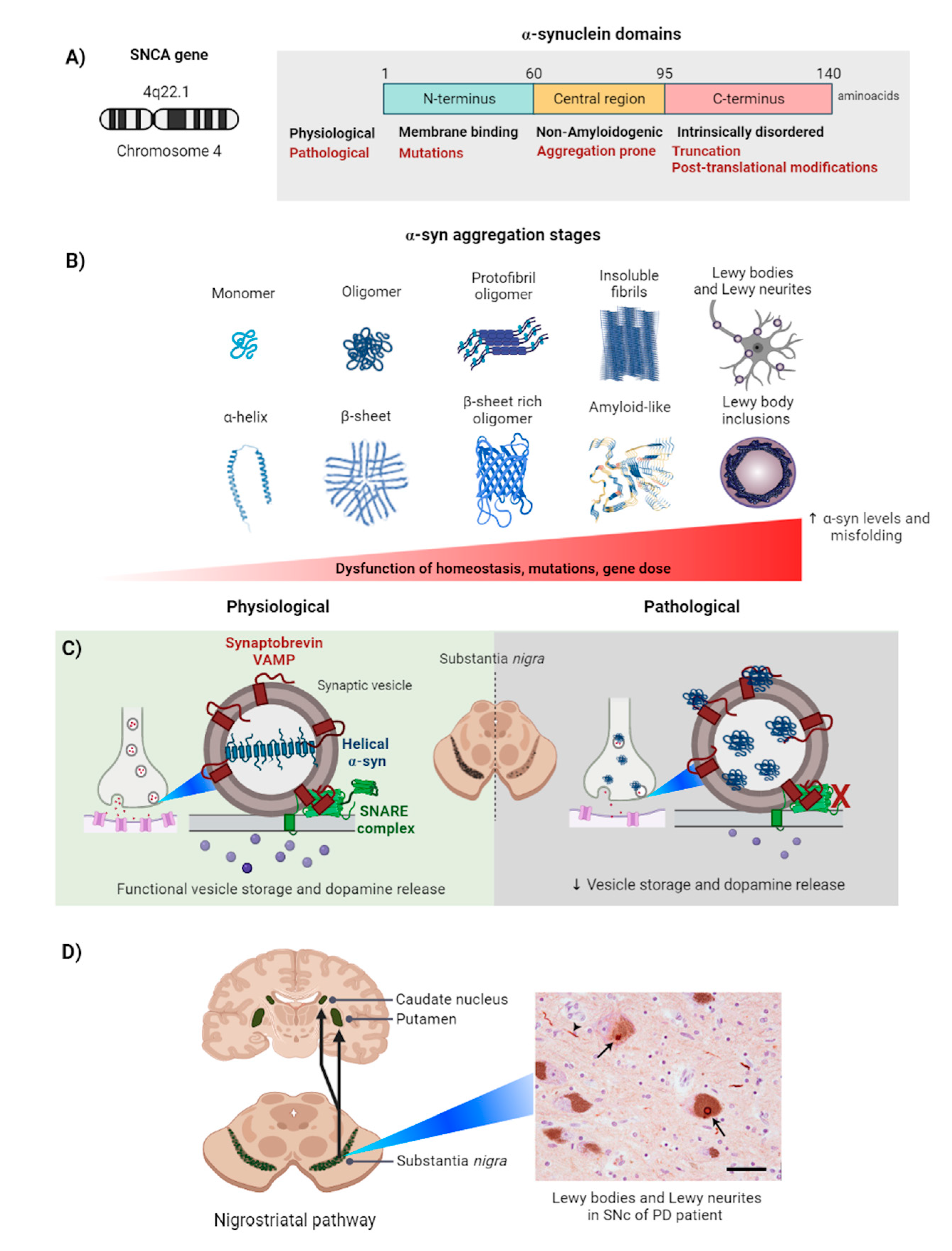
2. Alpha-Synuclein
2.1. Alpha-Synuclein Aggregation Induces Parkinson’s Disease
2.2. Alpha-Synuclein Aggregation in the Cell
2.3. Physiological Response to α-Syn Aggregation: Autophagy and Proteosomes
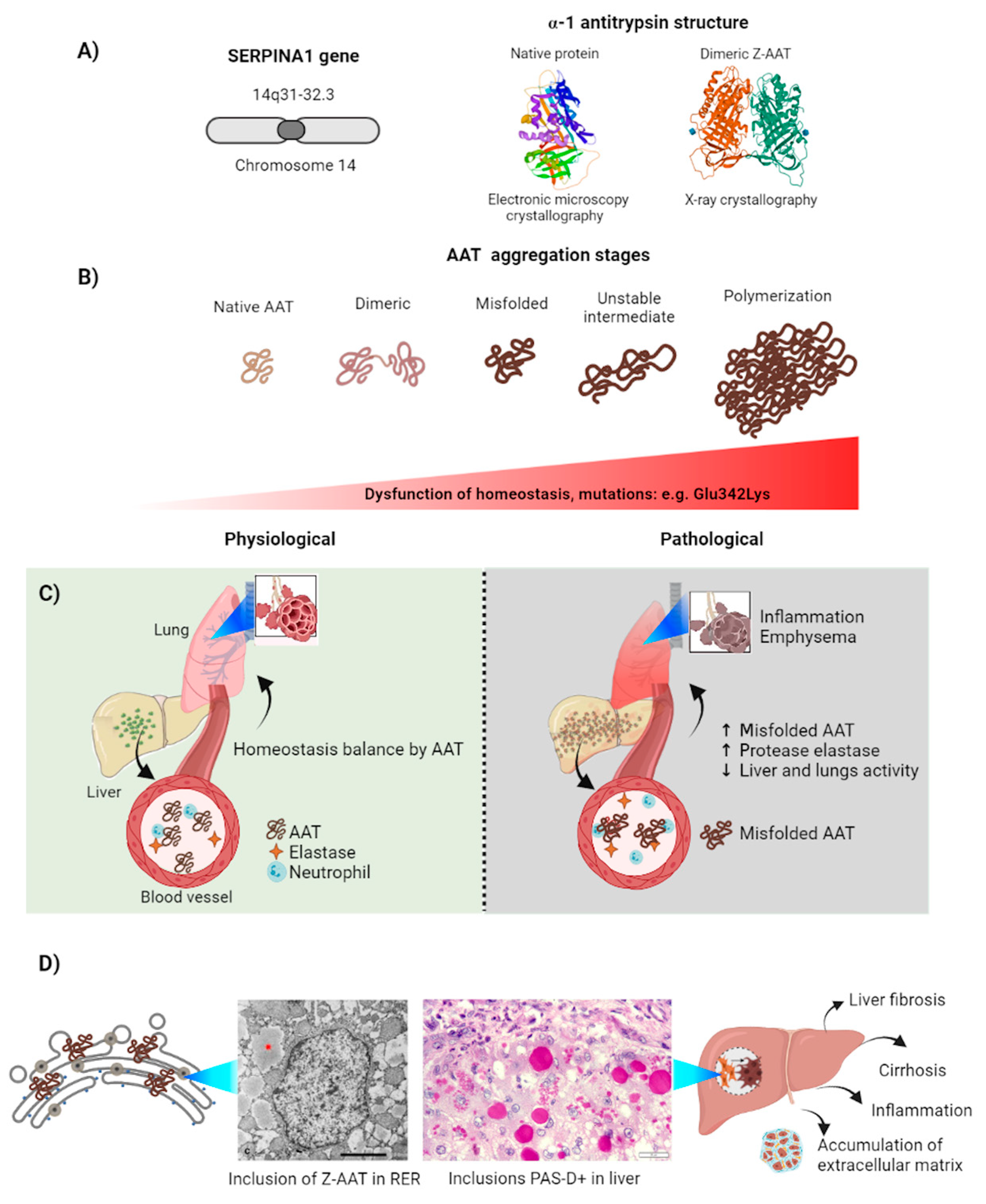
3. Alpha-1-Antitrypsin
3.1. Alpha-1-Antitrypsin Aggregation Induces Serpinopathies
3.2. Z-AAT Aggregation in the Cell
3.3. Physiological Response to Z-AAT Aggregation: Autophagy and Proteosomes
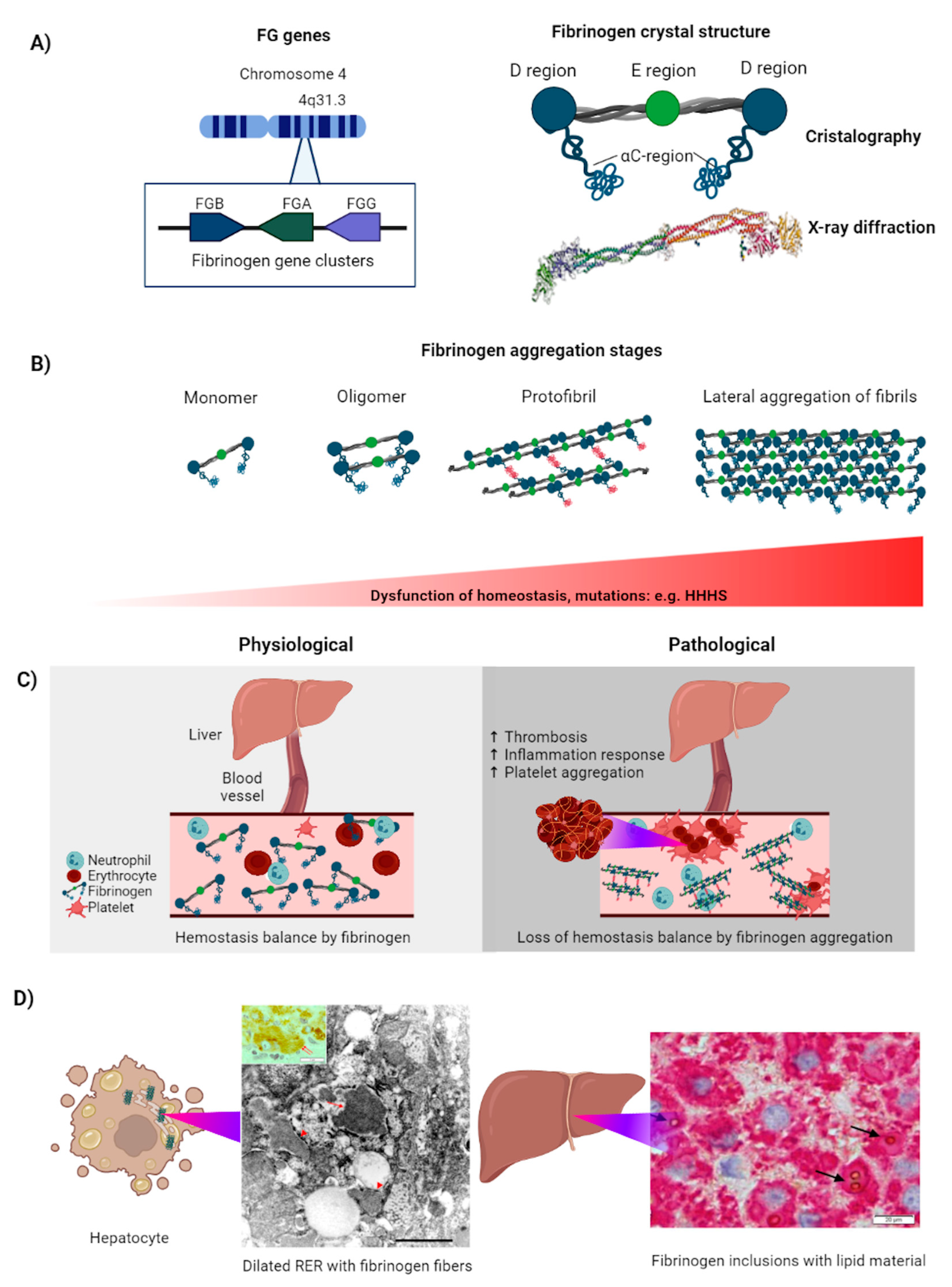
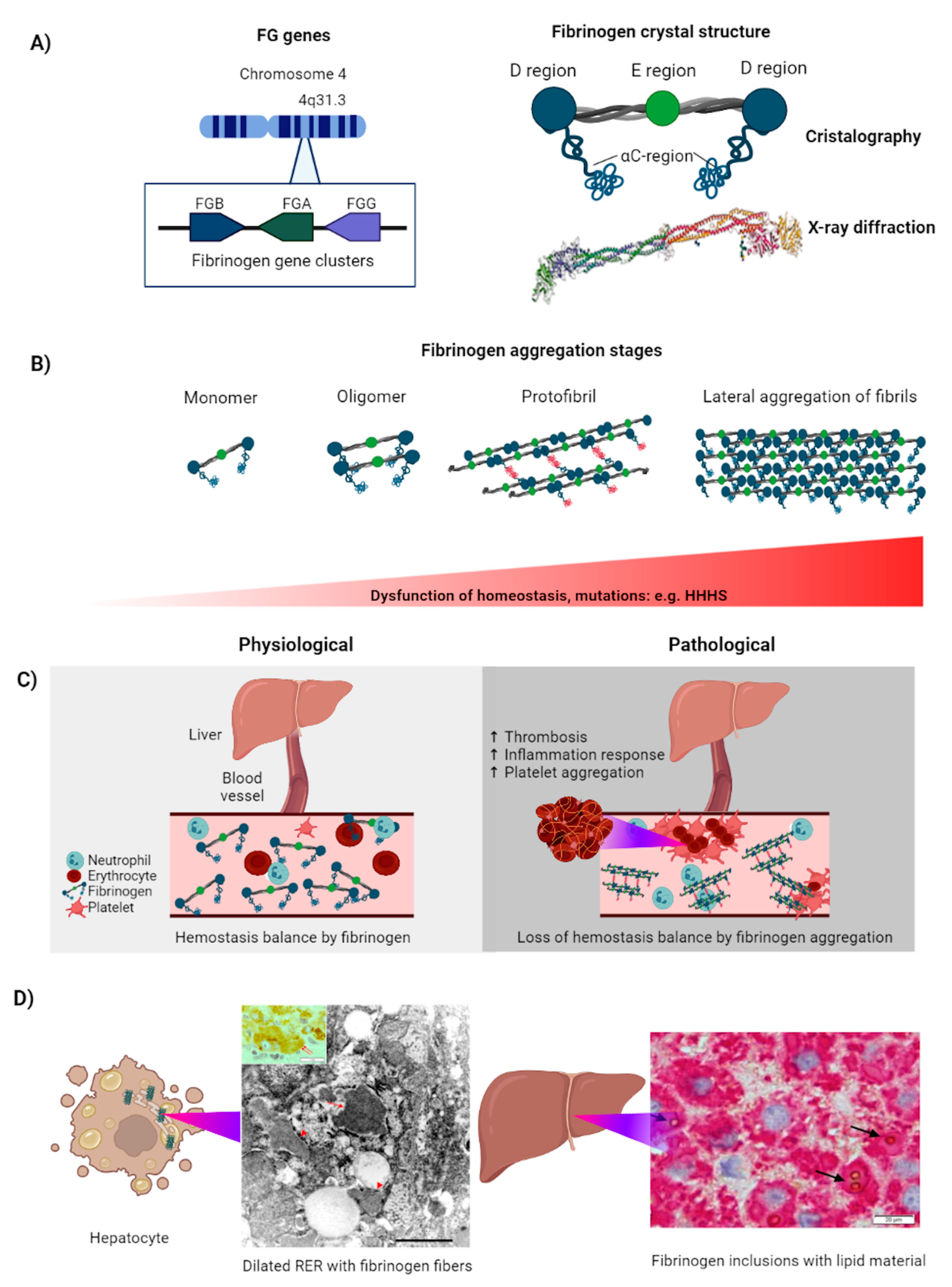
4. Fibrinogen
4.1. Fibrinogen Aggregation Induces Coagulopathies
4.2. FG Aggregation in the Cell
4.3. Physiological Response to FG Aggregation: Autophagy and Proteosomes
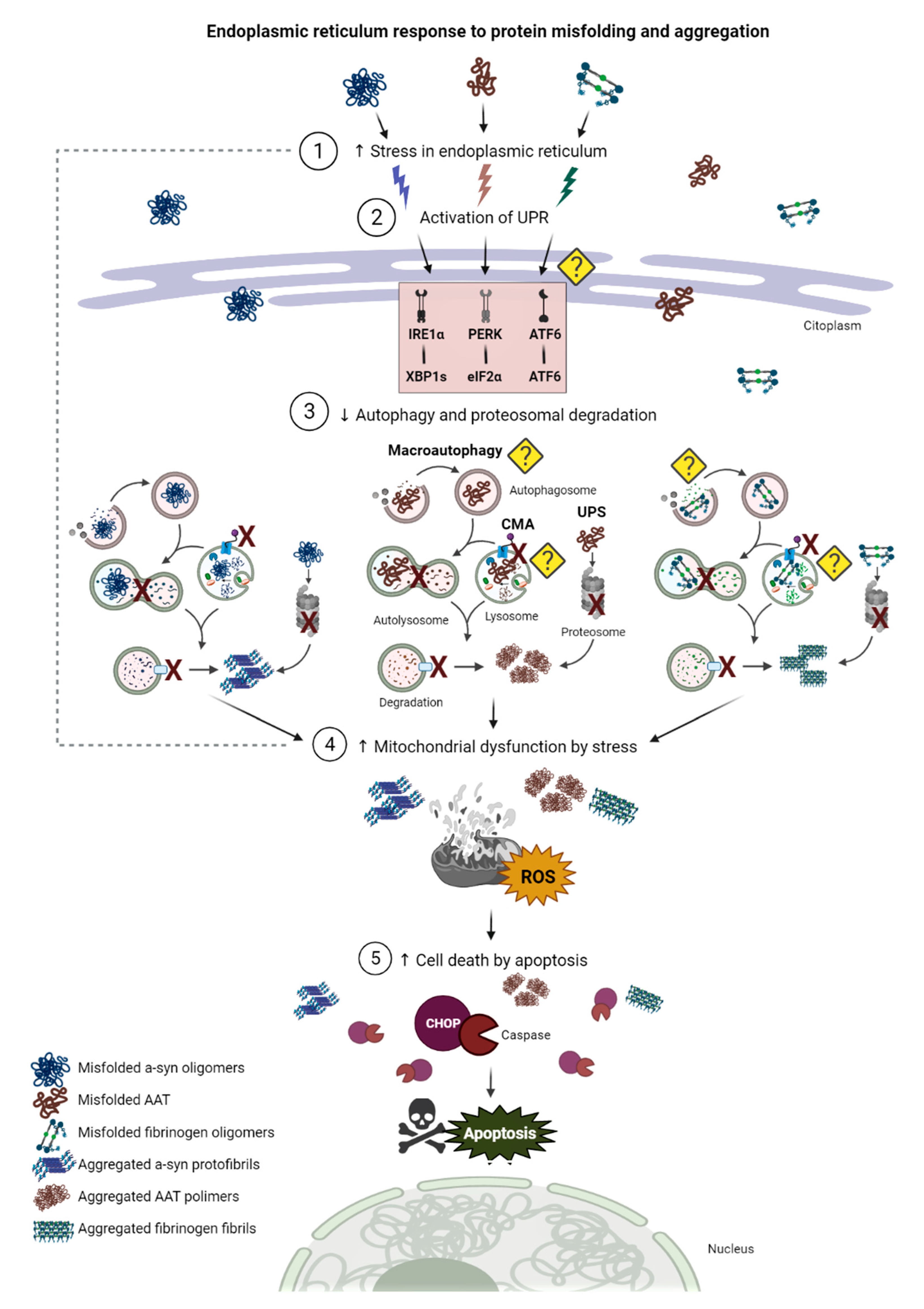
5. Endoplasmic Reticulum Stress and Unfolded Protein Response
5.1. The ER and the ER Stress
5.2. The Unfolded Protein Response
5.3. ER Stress and UPR in PD
5.4. ER Stress and UPR in AATD
5.5. ER Stress and UPR in HHHS
6. Hallmark Findings Comparison
7. Clinical Perspectives
7.1. Proteolytic Pathways Induction as Potential Treatment for α-Syn Aggregation in PD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parkinson’s Disease | ||||
|---|---|---|---|---|
| Target | Strategy | Results * | Conclusions | Ref. |
| ER stress | Transgenic mice over-expressing WT or mutant (A53T and A30P) α-syn treated with Salubrinal | ↓ α-syn oligomer ↓ ER stress | α -syn oligomers cause neurodegeneration by chronic ER stress response in vivo | [171] |
| ER stress | Mithramycin (MTM) administration in organotypic hippocampal slice cultures | ↓ ER stress-induced neurotoxicity ↓ Cell death by CHOP inhibition | MTM is a protective agent against ER stress neuronal death in vitro | [214] |
| ER stress | Tangeretin administration in mice injected with tunicamycin | ↑ Expression of GRP78 in SNpc ↓ Cell death induced by MPTP | Tangeretin regulates ER stress-related to PD | [223] |
| ER stress and UPR | Genetic deletion of ATF6α in transgenic mice treated with MPTP | ↓ TH levels and ↓ Number of dopaminergic neurons in SNpc | ATF6α exerts neuroprotection of dopaminergic neurons from MPTP toxicity in vivo | [218] |
| ER stress and UPR | Mouse model with deletion of ATF6α gene and injection of MPTP and probenecid (MPTP/P) | ↓ GRP78 ↑ Neuronal degeneration ↑ Ubiquitin accumulation ↓ Astroglial activation ↓ BDNF ↓ Anti-oxidative genes ↓ CHOP | UPR is activated in a model of chronic MPTP/P injection causing neurodegeneration | [219] |
| ER stress and UPR | Administration of tangeretin into mice with deletion of ATF6α and MPTP/P | ↑ UPR-target genes ↑ Dopaminergic neuronal survival ↑ Astrocyte survival | UPR contributes to the survival of dopaminergic neurons in SNpc | [219] |
| ER stress and UPR | Overexpression of chaperones GRP78/BiP in α-syn rat model of PD | ↓ α-syn neurotoxicity ↓ Apoptosis in TH neurons of SNpc ↑ Levels of striatal dopamine release | The GRP78/BiP plays a neuroprotective role in α-syn neurodegeneration | [220] |
| Macroautophagy | Overexpression of α-syn in cell cultures (SKNSH, HeLa and HEK293 lines) | ↑ p62 and ↓ LC3-II ↓ RAB1 homeostasis ↓ Omegasome formation Mislocalization of ATG-9 | Rab1a, α-syn, and ATG-9 regulate the formation of Omegasome | [60] |
| Autophagy–lysosome system | Overexpression of α-syn by lentivirus transduction and co-expression of Beclin-1 in a neuronal cell line | ↓ Accumulation of α-syn ↓ Neuritic alterations ↑ Effects of Beclin-1 by Rapamycin ↑ Lysosomal activation ↓ Synaptic and dendritic pathology ↓ Alterations in autophagy pathway | Beclin-1 decreases neuronal pathology of α-syn by inducing autophagy in vitro | [174] |
| Macroautophagy | Induction of macroautophagy by administration of trehalose in A53T α-syn transgenic rats | ↓ α-syn accumulation and aggregation in SNpc ↓ α-syn deficits in motor asymmetry ↑ Survival of dopaminergic neurons ↑ Dopamine turnover | Induction of macroutophagy prevents/ reverse α-syn aggregation in models of PD | [230] |
| CMA | Overexpression of LAMP2A in SH-SY5Y cells, rat cortical neurons in vitro, and SNpc neurons in vivo | ↓ α-syn neurotoxicity ↑ Survival of SNpc dopaminergic neurons ↑ Functionality of dopaminergic striatal terminals | Induction of CMA provide a novel therapeutic strategy for treatment of PD | [232] |
| Autophagy–lysosome system | Overexpressing of GCase in A53T α-syn transgenic mice | ↓ Soluble α-syn levels | GCase represents a potential therapeutic strategy for PD | [233] |
7.2. Proteolytic Pathways Induction as Potential Treatment for AAT Aggregation in AATD
| α-1-Antitrypsin Deficiency | ||||
|---|---|---|---|---|
| Target | Strategy | Results * | Conclusions | Ref. |
| Block polymerization of Z-AAT | Administration of 6-Mer reactive loop peptide (FLEAIG) | ↓ Polymerization of Z- AAT | Small molecule inhibitors can be used to treat Z-AAT deficiency. | [245] |
| ER stress and UPR | Administration of modulators of UPR: Sarcosine, Betaine, Hydroxyectoine and Ectoine in ER-stress induced by Tunicamycin | ↑ Restoration of homeostasis ↓ Levels of GRP78 and ATF-4 | Modulators of UPR mitigate the pathophysiological state of ER-stress. | [246] |
| Reverse misfolding of AAT | Administration of chemical chaperone: 4-phenylbutyric acid (PBA) in cell culture system and Z-AAT mice | ↓ Z-AAT secretion levels in cell culture and murine models | PBA is an important treatment of target organ injury in AAT deficiency | [247] |
| Polymerization of Z-AAT | Administration of trimethylamine N-oxide (TMAO) | ↓ Conversion of the native state to a polymerogenic intermediate | TMAO control the conformational transitions of folded AAT | [248] |
| Autophagy | Administration of autophagy enhancing drug carbamazepine (CBZ) in HeLa cell line HTO/Z and ATG-5–deficient cell line | ↓ Levels of ATZ insoluble and soluble fractions ↑ Autophagic flux by LC3-I and LC3-II ↓ Levels of soluble and insoluble ATZ in ATG-5 deficient line | CBZ is efficient in AAT deficiency as autophagy enhancer. | [110] |
| Autophagy | Activation of ATF6 by expression of spliced ATF6 (1–373 exons) | ↑ ER-associated degradation of Z-AAT ↓ Hepatocyte loss | ATF6 pathway limits Z-AAT cell toxicity | [251] |
| Autophagy | Cell lines (mouse embryonic fibroblast) with deletion in ATG-5 gene | ↓ Degradation of Z-AAT ↑ Z-AAT inclusions | Autophagic degradation prevent toxic accumulation of Z-AAT. | [235] |
| Autophagy | Effect of rapamycin on mouse model of Z-AAT | ↑ Autophagic activity by number of vacuoles ↓ Intrahepatic accumulation of Z-AAT ↓ Caspase 12 levels ↓ Hepatic fibrosis | Rapamycin reduces polymerized Z-AAT and progression of liver injury. | [236] |
| Autophagy | Liver-directed gene transfer of transcription factor EB (TFEB) in a mouse model of SERPINA1 deficiency. | ↓ Expression of SERPINA1 monomer ↑ Degradation of SERPINA1 polymer by autolysosomes ↓ Apoptosis and fibrosis 236 | TFEB gene transfer is a novel strategy for liver disease in SERPINA1 deficiency and prevent accumulation of toxic proteins. | [237] |
7.3. Proteolytic Pathways Induction as Potential Treatment for FG Aggregation in HHHS
7.4. Future Research through a Simplified Approach
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; George, G.; Mori, K. Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycoproteins and non-glycoproteins. Biochim. Biophys. Acta Gen. Subj. 2020, 1865, 129812. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteomics 2019, 1867, 890–908. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, T.K.; Paul, S. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J. 2006, 273, 1331–1349. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Gilbert, R.M. Epidemiology of Parkinson Disease. Neurol. Clin. 2016, 34, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein Misfolding Diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V.; Andreola, A.; Palladini, G.; Obici, L.; Casarini, S.; Perfetti, V. Protein Aggregation. Clin. Chem. Lab. Med. 2001, 39, 1065–1075. [Google Scholar] [CrossRef]
- Ferreira, N.d.C.; Caughey, B. Proteopathic Seed Amplification Assays for Neurodegenerative Disorders. Clin. Lab. Med. 2020, 40, 257–270. [Google Scholar] [CrossRef]
- Bendor, J.; Logan, T.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar] [CrossRef]
- Lima, V.d.A.; do Nascimiento, L.A.; Eliezer, D.; Follmer, C. Role of Parkinson’s Disease-Linked Mutations and N-Terminal Acetylation on the Oligomerization of α-Synuclein Induced by 3,4-Dihydroxyphenylacetaldehyde. ACS Chem. Neurosci. 2019, 10, 690–703. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, F.; Ventura, S. Inducing α-synuclein compaction: A new strategy for inhibiting α-synuclein aggregation? Neural Regen. Res. 2019, 14, 1897. [Google Scholar] [CrossRef]
- Sorrentino, Z.; Giasson, B.I. The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhena, F.; Faustini, G.; Bellucci, A. Study of alpha-synuclein fibrillation: State of the art and expectations. Neural Regen. Res. 2020, 15, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Ingelsson, M. Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other lewy body disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breydo, L.; Wu, J.W.; Uversky, V.N. A-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovere, M.; Sanderson, J.B.; Fonseca-Ornelas, L.; Patel, D.S.; Bartels, T. Refolding of helical soluble α-synuclein through transient interaction with lipid interfaces. FEBS Lett. 2018, 592, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.T.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Südhof, T.C. A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef]
- Peelaerts, W.; Baekelandt, V. ɑ-Synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 2016, 139, 256–274. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Delamarre, A.; Meissner, W.G. Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Press. Médicale 2017, 46, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Klein, C. The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons. Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, A.; Gonçalves, N.P.; Vægter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Del Mar, C.; Greenbaum, E.A.; Mayne, L.; Englander, S.W.; Woods, V.L., Jr. L. Structure and properties of alpha-synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. USA 2005, 102, 15477–15482. [Google Scholar] [CrossRef] [Green Version]
- Gurry, T.; Ullman, O.; Fisher, C.K.; Perovic, I.; Pochapsky, T.; Stultz, C.M. The dynamic structure of α-synuclein multimers. J. Am. Chem. Soc. 2013, 135, 3865–3872. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Lil, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Roostaee, A.; Beaudoin, S.; Staskevicius, A.; Roucou, X. Aggregation and neurotoxicity of recombinant α-synuclein aggregates initiated by dimerization. Mol. Neurodegener. 2013, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Singh, Y.; Sharpe, P.C.; Hoang, H.N.; Lucke, A.J.; McDowall, A.W.; Bottomley, S.P.; Fairlie, D.P. Amyloid formation from an α-helix peptide bundle is seeded by 3(10)-helix aggregates. Chemistry (Easton) 2011, 17, 151–160. [Google Scholar] [CrossRef]
- Stephens, A.D.; Zacharopoulou, M.; Kaminski Schierle, G.S. The Cellular Environment Affects Monomeric α-Synuclein Structure. Trends Biochem. Sci. 2019, 44, 453–466. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 2020, 134, 104623. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, P.; Yue, Z. Deregulation of autophagy and vesicle trafficking in Parkinson’s disease. Neurosci. Lett. 2019, 697, 59–65. [Google Scholar] [CrossRef]
- Bellomo, G.; Paciotti, S.; Gatticchi, L.; Parnetti, L. The vicious cycle between α-synuclein aggregation and autophagic-lysosomal dysfunction. Mov. Disord. 2020, 35, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [Green Version]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.; Brunden, K.R.; Lee, V.M.Y. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Liang, Y.; Schools, S.; Dawson, V.L.; Dawson, T.M.; Savitt, J.M. Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J. Neurosci. 2012, 32, 16503–16509. [Google Scholar] [CrossRef]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [Green Version]
- Maday, S.; Holzbaur, E.L.F. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 2014, 30, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.-T.; Portes Ureshino, R.; et al. Overexpression of α-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Olsen, A.L.; Sygnecka, K.; Lohr, K.M.; Feany, M.B. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet. 2021, 17, e1009359. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Reimão-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhães, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef] [Green Version]
- Shahpasandzadeh, H.; Popova, B.; Kleinknecht, A.; Fraser, P.E.; Outeiro, T.F.; Braus, G.H. Interplay between sumoylation and phosphorylation for protection against α-synuclein inclusions. J. Biol. Chem. 2014, 289, 31224–31240. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and alpha-synuclein: Relevance to Parkinson’s disease and related synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 2000, 113, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant α-Synuclein Confers Toxicity to Neurons in Part through Inhibition of Chaperone-Mediated Autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, J.A.; Angot, E.; Brundin, P. A deadly spread: Cellular mechanisms of α-synuclein transfer. Cell Death Differ. 2011, 18, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- Sacino, A.N.; Brooks, M.M.; Chakrabarty, P.; Saha, K.; Khoshbouei, H.; Golde, T.E.; Giasson, B.I. Proteolysis of α-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J. Neurochem. 2017, 140, 662–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T.; Rossi, A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef]
- Parnetti, L.; Farotti, L.; Eusebi, P.; Chiasserini, D.; De Carlo, C.; Giannandrea, D.; Salvadori, N.; Lisetti, V.; Tambasco, N.; Rossi, A.; et al. Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson’s Disease. Front. Aging Neurosci. 2014, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Yang, N.Y.; Lee, C.; Lee, H.-J.; Kim, S.; Sardi, S.P.; Lee, S.-J. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, S.; Kam, T.-I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Wolf, P.; Levy, O.A.; Kang, U.J.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.H.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yan, B.; Wang, X.; Feng, X.; Zhang, A.; Xu, X.; Dong, H. Decreased activities of lysosomal acid alpha-D-galactosidase A in the leukocytes of sporadic Parkinson’s disease. J. Neurol. Sci. 2008, 271, 168–173. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Jenner, P.; Olanow, C.W.; Isacson, O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- Bentea, E.; Van der Perren, A.; Liefferinge, J.V.; El Arfani, A.; Albertini, G.; Demuyser, T.; Merckx, E.; Michotte, Y.; Smolders, I.; Baekelandt, V.; et al. Nigral proteasome inhibition in mice leads to motor and non-motor deficits and increased expression of Ser129 phosphorylated α-synuclein. Front. Behav. Neurosci. 2015, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zheng, Y.; Zhang, F.; Wei, Z.; Wang, Y.; Carrell, R.W.; Read, R.J.; Chen, G.Q.; Zhou, A. Molecular mechanism of Z α1-antitrypsin deficiency. J. Biol. Chem. 2016, 291, 15674–15686. [Google Scholar] [CrossRef] [Green Version]
- Laffranchi, M.; Elliston, E.L.; Miranda, E.; Perez, J.; Ronzoni, R.; Jagger, A.M.; Heyer-Chauhan, N.; Brantly, M.L.; Fra, A.; Lomas, D.A.; et al. Intrahepatic heteropolymerization of M and Z alpha-1-antitrypsin. JCI insight 2020, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z α1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Kolarich, D.; Weber, A.; Turecek, P.L.; Schwarz, H.P.; Altmann, F. Comprehensive glyco-proteomic analysis of human α1- antitrypsin and its charge isoforms. Proteomics 2006, 6, 3369–3380. [Google Scholar] [CrossRef]
- Karatas, E.; Di-tommaso, S.; Dugot-senant, N. Overview of Alpha-1 Antitrypsin Deficiency- Mediated Liver Disease. Eur. Med. J. 2019, 7, 65–79. [Google Scholar]
- Roussel, B.D.; Irving, J.A.; Ekeowa, U.I.; Belorgey, D.; Haq, I.; Ordóñez, A.; Kruppa, A.J.; Duvoix, A.; Rashid, S.T.; Crowther, D.C.; et al. Unravelling the twists and turns of the serpinopathies. FEBS J. 2011, 278, 3859–3867. [Google Scholar] [CrossRef] [PubMed]
- Matamala, N.; Martínez, M.T.; Lara, B.; Pérez, L.; Vázquez, I.; Jiménez, A.; Barquín, M.; Ferrarotti, I.; Blanco, I.; Janciauskiene, S.; et al. Alternative transcripts of the SERPINA1 gene in alpha-1 antitrypsin deficiency. J. Transl. Med. 2015, 13, 211. [Google Scholar] [CrossRef] [Green Version]
- Callea, F.; Francalanci, P.; Giovannoni, I. Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases. Int. J. Mol. Sci. 2021, 22, 5778. [Google Scholar] [CrossRef]
- Lawless, M.W.; Mankan, A.K.; Gray, S.G.; Norris, S. Endoplasmic reticulum stress--a double edged sword for Z alpha-1 antitrypsin deficiency hepatoxicity. Int. J. Biochem. Cell Biol. 2008, 40, 1403–1414. [Google Scholar] [CrossRef]
- Silverman, E.K.; Sandhaus, R.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2009, 360, 2749–2757. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. α1-Antitrypsin deficiency. Nat. Rev. Dis. Prim. 2016, 2, 16051. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi∗Z variant increases the risk to develop liver cirrhosis. Gut 2019, 68, 1099–1107. [Google Scholar] [CrossRef]
- Edgar, R.G.; Patel, M.; Bayliss, S.; Crossley, D.; Sapey, E.; Turner, A.M. Treatment of lung disease in alpha-1 antitrypsin deficiency: A systematic review. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 1295–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhaus, R.A.; Turino, G.; Brantly, M.L.; Campos, M.; Cross, C.E.; Goodman, K.; Hogarth, D.K.; Knight, S.L.; Stocks, J.M.; Stoller, J.K.; et al. The Diagnosis and Management of Alpha-1 Antitrypsin Deficiency in the Adult. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 3, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callea, F.; Brisiptti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J. Hepatic endoplasmic reticulum storage diseases. Liver 2008, 12, 357–362. [Google Scholar] [CrossRef]
- Dickens, J.A.; Ordóñez, A.; Chambers, J.E.; Beckett, A.J.; Patel, V.; Malzer, E.; Dominicus, C.S.; Bradley, J.; Peden, A.A.; Prior, I.A.; et al. The endoplasmic reticulum remains functionally connected by vesicular transport after its fragmentation in cells expressing Z-α1-antitrypsin. FASEB J. 2016, 30, 4083–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faull, S.V.; Elliston, E.L.K.; Gooptu, B.; Jagger, A.M.; Aldobiyan, I.; Redzej, A.; Badaoui, M.; Heyer-Chauhan, N.; Rashid, S.T.; Reynolds, G.M.; et al. The structural basis for Z α1-antitrypsin polymerization in the liver. Sci. Adv. 2020, 6, eabc1370. [Google Scholar] [CrossRef]
- Elliott, P.R.; Pei, X.Y.; Dafforn, T.R.; Lomas, D.A. Topography of a 2.0 Å structure of α 1 -antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 2000, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.A.; Sendall, T.J.; Pedersen, J.S.; Kjeldgaard, M.; Huntington, J.A.; Jensen, J.K. The shapes of Z-α1-antitrypsin polymers in solution support the c-terminal domain-swap mechanism of polymerization. Biophys. J. 2014, 107, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, A.S.; Perlmutter, D.H.; Wang, Y. Capitalizing on the autophagic response for treatment of liver disease caused by alpha-1-antitrypsin deficiency and other genetic diseases. Biomed Res. Int. 2014, 2014, 459823. [Google Scholar] [CrossRef] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Mukherjee, A.; Hidvegi, T.; Araya, P.; Ewing, M.; Stolz, D.B.; Perlmutter, D.H. NFκB mitigates the pathological effects of misfolded α1-antitrypsin by activating autophagy and an integrated program of proteostasis mechanisms. Cell Death Differ. 2019, 26, 455–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cobanoglu, M.C.; Li, J.; Hidvegi, T.; Hale, P.; Ewing, M.; Chu, A.S.; Gong, Z.; Muzumdar, R.; Pak, S.C.; et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded α1-antitrypsin Z. PLoS ONE 2019, 14, e0209748. [Google Scholar] [CrossRef]
- Fan, J.; Shi, Y.; Peng, Y. Autophagy and liver diseases. Adv. Exp. Med. Biol. 2020, 1207, 497–528. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-Associated Phagocytosis and Inflammation. J. Mol. Biol. 2017, 429, 3561–3576. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, J.; Zhu, N.; Ding, Q.; Zhang, X.; Yu, J.; Qiang, W.; Zhang, Z.; Ma, Y.; Huang, D.; et al. Ubiquitin ligase SYVN1/HRD1 facilitates degradation of the SERPINA1 Z variant/α-1-antitrypsin Z variant via SQSTM1/p62-dependent selective autophagy. Autophagy 2017, 13, 686–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Blomenkamp, K.S.; Fickert, P.; Trauner, M.; Teckman, J.H. NorUDCA promotes degradation of α1-antitrypsin mutant Z protein by inducing autophagy through AMPK/ULK1 pathway. PLoS One 2018, 13, e0200897. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Lin, S.C. The core domain of RGS16 retains G-protein binding and GAP activity in vitro, but is not functional in vivo. FEBS Lett. 1998, 422, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Wauson, E.M.; Dbouk, H.A.; Ghosh, A.B.; Cobb, M.H. G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 2014, 25, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Park, J.; Kim, M.S.; Ha, J.Y.; Jang, Y.W.; Shin, D.H.; Son, J.H. The Tnfaip8-PE complex is a novel upstream effector in the anti-autophagic action of insulin. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Hidvegi, T.; Mirnics, K.; Hale, P.; Ewing, M.; Beckett, C.; Perlmutter, D.H. Regulator of G signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant α1- antitrypsin Z in the classical form of α1-antitrypsin deficiency. J. Biol. Chem. 2007, 282, 27769–27780. [Google Scholar] [CrossRef] [Green Version]
- Karatas, E.; Bouchecareilh, M. Alpha 1-antitrypsin deficiency: A disorder of proteostasis-mediated protein folding and trafficking pathways. Int. J. Mol. Sci. 2020, 21, 1493. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1?SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Qu, D.; Teckman, J.H.; Omura, S.; Perlmutter, D.H. Degradation of a mutant secretory protein, α 1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol. Chem. 1996, 271, 22791–22795. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Q.; Shen, Y.; Sun, A.; Zhu, X.; Fang, S.; Shen, Y. The ubiquitin ligase Hrd1 promotes degradation of the Z variant alpha 1-antitrypsin and increases its solubility. Mol. Cell. Biochem. 2011, 346, 137–145. [Google Scholar] [CrossRef]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: One for soluble Z variant of human α-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell 2006, 17, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burcu, G.; Bellacchio, E.; Sag, E.; Cebi, A.H.; Saygin, I.; Bahadir, A.; Yilmaz, G.; Corbeddu, M.; Cakir, M.; Callea, F. Structural characteristics in the γ chain variants associated with fibrinogen storage disease suggest the underlying pathogenic mechanism. Int. J. Mol. Sci. 2020, 21, 5139. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.M.; Duga, S. Hereditary Hypofibrinogenemia with Hepatic Storage. J. Mol. Sci 2020, 21, 7830. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wang, B.; Liu, L.; Gan, Q.; Liu, X.; Chen, L.; Chen, L. Hepatic fibrinogen storage disease and hypofibrinogenemia caused by fibrinogen Aguadilla mutation: A case report. J. Int. Med. Res. 2020, 48, 030006051989803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neerman-Arbez, M.; Antonarakis, S.E.; Honsberger, A.; Morris, M.A. The 11 kb FGA deletion responsible for congenital afibrinogenaemia is mediated by a short direct repeat in the fibrinogen gene cluster. Eur. J. Hum. Genet. EJHG 1999, 7, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Callea, F.; Desmet, V. The discovery of endoplasmic reticulum storage disease. The connection between an h&e slide and the brain. Int. J. Mol. Sci. 2021, 22, 2899. [Google Scholar] [CrossRef]
- Dib, N.; Quelin, F.; Ternisien, C.; Hanss, M.; Michalak, S.; De Mazancourt, P.; Rousselet, M.C.; Calès, P. Fibrinogen angers with a new deletion (γ GVYYQ 346-350) causes hypofibrinogenemia with hepatic storage. J. Thromb. Haemost. 2007, 5, 1999–2005. [Google Scholar] [CrossRef]
- Asselta, R.; Robusto, M.; Braidotti, P.; Peyvandi, F.; Nastasio, S.; D’Antiga, L.; Perisic, V.N.; Maggiore, G.; Caccia, S.; Duga, S. Hepatic fibrinogen storage disease: Identification of two novel mutations (p.Asp316Asn, fibrinogen Pisa and p.Gly366Ser, fibrinogen Beograd) impacting on the fibrinogen γ-module. J. Thromb. Haemost. 2015, 13, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Gettins, P.G. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Sari, S.; Gulda, E.; Dalgic, B.; Akyol, G.; Sogo, T.; Al-Hussaini, A.; Maggiore, G.; Bartuli, A.; et al. Fibrinogen gamma chain mutations provoke fibrinogen and apolipoprotein B plasma deficiency and liver storage. Int. J. Mol. Sci. 2017, 18, 2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Redman, C. The Degradation of Nascent Fibrinogen Chains Is Mediated by the Ubiquitin Proteasome Pathway. Biochem. Biophys. Res. Commun. 1999, 261, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.M.; Xia, H. Fibrinogen biosynthesis. Assembly, intracellular degradation, and association with lipid synthesis and secretion. Ann. N. Y. Acad. Sci. 2001, 936, 480–495. [Google Scholar] [CrossRef]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Autophagy: An ER protein quality control process. Autophagy 2006, 2, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Le Fourn, V.; Park, S.; Jang, I.; Gaplovska-Kysela, K.; Guhl, B.; Lee, Y.; Won Cho, J.; Zuber, C.; Roth, J. Large protein complexes retained in the ER are dislocated by non-COPII vesicles and degraded by selective autophagy. Cell. Mol. Life Sci. 2013, 70, 1985–2002. [Google Scholar] [CrossRef] [Green Version]
- Puls, F.; Goldschmidt, I.; Bantel, H.; Agne, C.; Bröcker, V.; Dämmrich, M.; Lehmann, U.; Berrang, J.; Pfister, E.-D.; Kreipe, H.H.; et al. Autophagy-enhancing drug carbamazepine diminishes hepatocellular death in fibrinogen storage disease. J. Hepatol. 2013, 59, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- da Costa, C.A.; El Manna, W.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Hassler, J.R.; Scheuner, D.L.; Wang, S.; Han, J.; Kodali, V.K.; Li, P.; Nguyen, J.; Goerge, J.S.; Davis, C.; Wu, S.P.; et al. The IRE1α/XBP1s Pathway Is Essential for the Glucose Response and Protection of β Cells. PLoS Biol. 2015, 13, e1002277. [Google Scholar] [CrossRef] [Green Version]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci. 2002, 22, 10690–10698. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.J.M.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Esteves, A.R.; Cardoso, S.M. Differential protein expression in diverse brain areas of Parkinson’s and Alzheimer’s disease patients. Sci. Rep. 2020, 10, 13149. [Google Scholar] [CrossRef]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Turano, C.; Coppari, S.; Altieri, F.; Ferraro, A. Proteins of the PDI family: Unpredicted non-ER locations and functions. J. Cell. Physiol. 2002, 193, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Heman-Ackah, S.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.; Lee, G.H.; Li, B.; Park, S.A.; Lee, S.-J.; Chae, H.-J. Endoplasmic reticulum stress induced by manganese trigger a-synuclein accumulation. Trop. J. Off Pharm. Res. 2018, 17, 1497–1503. [Google Scholar] [CrossRef]
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of α-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23178–23188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gan, M.; Ebrahim, A.S.; Lin, W.-L.; Melrose, H.L.; Yen, S.-H.C. ER stress response plays an important role in aggregation of α-synuclein. Mol. Neurodegener. 2010, 5, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [Green Version]
- Paiva, I.; Jain, G.; Lázaro, D.F.; Jerčić, K.G.; Hentrich, T.; Kerimoglu, C.; Pinho, R.; Szegő, È.M.; Burkhardt, S.; Capece, V.; et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol. Dis. 2018, 119, 129–135. [Google Scholar] [CrossRef]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef]
- Ren, C.; Sun, K.; Zhang, Y.; Hu, Y.; Hu, B.; Zhao, J.; He, Z.; Ding, R.; Wang, W.; Liang, C. Sodium–Glucose CoTransporter-2 Inhibitor Empagliflozin Ameliorates Sunitinib-Induced Cardiac Dysfunction via Regulation of AMPK–mTOR Signaling Pathway–Mediated Autophagy. Front. Pharmacol. 2021, 12, 664181. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [Green Version]
- Hoang, S. a-Synuclein and Mitochondrial Dysfunction Induced ER Stress Leading to Parkinson’s Disease. Creat. Components 2020, 510, 1–24. [Google Scholar]
- Perlmutter, D.H. Liver injury in alpha1-antitrypsin deficiency: An aggregated protein induces mitochondrial injury. J. Clin. Invest. 2002, 110, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Teckman, J.H.; An, J.-K.; Loethen, S.; Perlmutter, D.H. Fasting in alpha1-antitrypsin deficient liver: Constitutive [correction of consultative] activation of autophagy. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Magallón, M.; Carrión, A.E.; Bañuls, L.; Pellicer, D.; Castillo, S.; Bondía, S.; Navarro-García, M.M.; González, C.; Dasí, F. Oxidative Stress and Endoplasmic Reticulum Stress in RareRespiratory Diseases. J. Clin. Med. 2021, 10, 1268. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.W.; Greene, C.M.; McLean, C.; Lawless, M.W.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of Bad. Hepatology 2007, 46, 496–503. [Google Scholar] [CrossRef]
- Kelly, E.; Greene, C.M.; Carroll, T.P.; McElvaney, N.G.; O’Neill, S.J. Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant alpha1-antitrypsin deficiency. J. Biol. Chem. 2009, 284, 16891–16897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar] [CrossRef] [Green Version]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, A.N.; O’Neill, S.J.; McElvaney, N.G. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J. Immunol. 2010, 184, 4538–4546. [Google Scholar] [CrossRef] [Green Version]
- Papp, E.; Száiraz, P.; Korcsmáiros, T.; Csermely, P. Changes of endoplasmic reticulum chaperone complexes, redox state, and impaired protein disulfide reductase activity in misfolding αi-antitrypsin transgenic mice. FASEB J. 2006, 20, 1018–1020. [Google Scholar] [CrossRef] [Green Version]
- Lawless, M.W.; Greene, C.M.; Mulgrew, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J. Immunol. 2004, 172, 5722–5726. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology 2012, 57, 2049–2060. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H.; Perlmutter, D.H. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; McElvaney, N.G. Z α-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Yu, S.; Banerjee, D.; Overton, O.; Mukhopadhyay, G.; Oddoux, C.; Grieninger, G.; Redman, C. Assembly and secretion of fibrinogen. Degradation of individual chains. J. Biol. Chem. 1992, 267, 23151–23158. [Google Scholar] [CrossRef]
- Huang, S.; Cao, Z.; Chung, D.W.; Davie, E.W. The role of betagamma and alphagamma complexes in the assembly of human fibrinogen. J. Biol. Chem. 1996, 271, 27942–27947. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Bae, E.-J.; Lee, S.-J. Extracellular α-synuclein—a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92–98. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhao, C.; Li, D.; Tian, Z.; Lai, Y.; Diao, J.; Liu, C. Versatile Structures of α-Synuclein. Front. Mol. Neurosci. 2016, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.Q.; Copray, S.J.C.V.M.; Ferrari, M.F.R. Alpha-synuclein toxicity on protein quality control, mitochondria and endoplasmic reticulum. Neurochem. Res. 2018, 43, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Nakata, Y.; Choong, C.J.; Mochizuki, H. Neurodegenerative changes initiated by presynaptic dysfunction. Transl. Neurodegener. 2013, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.J.; Lomas, D.A. Alpha 1 -Antitrypsin Deficiency and Autophagy. N. Engl. J. Med. 2010, 363, 1863–1864. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. The ER overload response: Activation of NF-κB. Trends Biochem. Sci. 1997, 22, 63–67. [Google Scholar] [CrossRef]
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in α 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Lindblad, D.; Blomenkamp, K.; Teckman, J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology 2007, 46, 1228–1235. [Google Scholar] [CrossRef]
- Al-Hussaini, A.; Altalhi, A.; El Hag, I.; Alhussaini, H.; Francalanci, P.; Giovannoni, I.; Callea, F. Hepatic fibrinogen storage disease due to the fibrinogen γ375 Arg → Trp mutation fibrinogen aguadilla is present in Arabs. Saudi J. Gastroenterol. 2014, 20, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Thangaraj, A.; Sil, S.; Tripathi, A.; Chivero, E.T.; Periyasamy, P.; Buch, S. Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 285–325. [Google Scholar] [CrossRef]
- Wei, J.; Fang, D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. [Google Scholar] [CrossRef]
- Venderova, K.; Park, D.S. Programmed Cell Death in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009365. [Google Scholar] [CrossRef]
- Erekat, N.S. Apoptosis and its Role in Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; pp. 65–82. [Google Scholar]
- John, A.E.; White, N.J. Platelets and fibrinogen: Emerging complexity in trauma-induced coagulopathy. Semin. Thromb. Hemost. 2020, 46, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Fewell, S.W.; Travers, K.J.; Weissman, J.S.; Brodsky, J.L. The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet. 2001, 35, 149–191. [Google Scholar] [CrossRef] [PubMed]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Shang, Y.; Tao, J.; Zhang, J.; Sha, B. Endoplasmic Reticulum Stress Signaling Pathways: Activation and Diseases. Curr. Protein Pept. Sci. 2019, 20, 935–943. [Google Scholar] [CrossRef]
- Bekker, M.; Abrahams, S.; Loos, B.; Bardien, S. Can the interplay between autophagy and apoptosis be targeted as a novel therapy for Parkinson’s disease? Neurobiol. Aging 2021, 100, 91–105. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, G.; Brostrom, M.A.; Brostrom, C.O. Demonstration of a calcium requirement for secretory protein processing and export. Differential effects of calcium and dithiothreitol. J. Biol. Chem. 1992, 267, 3932–3939. [Google Scholar] [CrossRef]
- Lodish, H.F.; Kong, N. Perturbation of cellular calcium blocks exit of secretory proteins from the rough endoplasmic reticulum. J. Biol. Chem. 1990, 265, 10893–10899. [Google Scholar] [CrossRef]
- Kosuge, Y.; Taniguchi, Y.; Imai, T.; Ishige, K.; Ito, Y. Neuroprotective effect of mithramycin against endoplasmic reticulum stress-induced neurotoxicity in organotypic hippocampal slice cultures. Neuropharmacology 2011, 61, 252–261. [Google Scholar] [CrossRef]
- Takano, K.; Tabata, Y.; Kitao, Y.; Murakami, R.; Suzuki, H.; Yamada, M.; Iinuma, M.; Yoneda, Y.; Ogawa, S.; Hori, O. Methoxyflavones protect cells against endoplasmic reticulum stress and neurotoxin. Am. J. Physiol. Cell Physiol. 2007, 292, C353–C361. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Huerta, P.; Troncoso-Escudero, P.; Jerez, C.; Hetz, C.; Vidal, R.L. The intersection between growth factors, autophagy and ER stress: A new target to treat neurodegenerative diseases? Brain Res. 2016, 1649, 137–180. [Google Scholar] [CrossRef]
- Egawa, N.; Yamamoto, K.; Inoue, H.; Hikawa, R.; Nishi, K.; Mori, K.; Takahashi, R. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem. 2011, 286, 7947–7957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashida, K.; Kitao, Y.; Sudo, H.; Awa, Y.; Maeda, S.; Mori, K.; Takahashi, R.; Iinuma, M.; Hori, O. ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS One 2012, 7, e47950. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyub, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010, 5, e9313. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Koprich, J.B.; Wang, Y.; Yu, W.; Xiao, B.; Brotchie, J.M.; Wang, J. Treatment with Trehalose Prevents Behavioral and Neurochemical Deficits Produced in an AAV α-Synuclein Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 2258–2268. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Kiosses, W.B.; Flynn, C.T.; Brady, N.R.; Fox, H.S. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One 2008, 3, e2906. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadoupoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.G.H.; Puyal, J. Autophagic cell death exists. Autophagy 2012, 8, 867–869. [Google Scholar] [CrossRef] [Green Version]
- Button, R.W.; Luo, S.; Rubinsztein, D.C. Autophagic activity in neuronal cell death. Neurosci. Bull. 2015, 31, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Kepp, O.; Kroemer, G. The end of autophagic cell death? Autophagy 2012, 8, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.F.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. α-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2020, 23, 21–31. [Google Scholar] [CrossRef]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Leon, C.; Bouchecareilh, M. The Autophagy Pathway: A Critical Route in the Disposal of Alpha 1-Antitrypsin Aggregates That Holds Many Mysteries. Int. J. Mol. Sci. 2021, 22, 1875. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.H.; Lee, K.N.; Kim, J. The Z type variation of human alpha 1-antitrypsin causes a protein folding defect. Nat. Struct. Biol. 1995, 2, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Mahadeva, R.; Dafforn, T.R.; Carrell, R.W.; Lomas, D.A. 6-merpeptide selectively anneals to a pathogenic serpin conformation and blocks po-lymerization: Implications for the prevention of Z_1-antitrypsin-related cirrhosis. J. Biol. Chem. 2002, 277, 6771–6774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazari, Y.M.; Habib, M.; Bashir, S.; Bashir, A.; Hilal, N.; Irfan, S.; Haq, E.U.; Fazili, K.M. Natural osmolytes alleviate GRP78 and ATF-4 levels: Corroboration for potential modulators of unfolded protein response. Life Sci. 2016, 146, 148–153. [Google Scholar] [CrossRef]
- Burrows, J.A.; Willis, L.K.; Perlmutter, D.H. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc. Natl. Acad. Sci. USA 2000, 97, 1796–1801. [Google Scholar] [CrossRef] [Green Version]
- Devlin, G.L.; Parfrey, H.; Tew, D.J.; Lomas, D.A.; Bottomley, S.P. Prevention of polymerization of M and Z alpha1-Antitrypsin (alpha1-AT) with trimethylamine N-oxide. Implications for the treatment of alpha1-at deficiency. Am. J. Respir. Cell Mol. Biol. 2001, 24, 727–732. [Google Scholar] [CrossRef]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Sundaram, A.; Appathurai, S.; Plumb, R.; Mariappan, M. Dynamic changes in complexes of IRE1alpha, PERK, and ATF6alpha during endoplasmic reticulum stress. Mol. Biol. Cell 2018, 29, 1376–1388. [Google Scholar] [CrossRef]
- Smith, S.E.; Granell, S.; Salcedo-Sicilia, L.; Baldini, G.; Egea, G.; Teckman, J.H.; Baldini, G. Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J. Biol. Chem. 2011, 286, 41563–41577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimoto, T.; Shoji, S.; Hidvegi, T.; Mizushima, N.; Umebayashi, K.; Perlmutter, D.H.; Yoshimori, T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006, 281, 4467–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washington University School of Medicine (U.S.). Carbamazepine in Severe Liver Disease Due to Alpha-1 Antitrypsin Deficiency (CBZ); U.S. National Library of Medicine: Bethesda, MA, USA, 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT01379469 (accessed on 5 October 2021).
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. (Maywood) 2010, 235, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Pastore, N.; Ballabio, A.; Brunetti-Pierri, N. Autophagy master regulator TFEB induces clearance of toxic SERPINA1/α-1-antitrypsin polymers. Autophagy 2013, 9, 1094–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellacchio, E. Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors. Int. J. Mol. Sci. 2020, 21, 9016. [Google Scholar] [CrossRef]
- Maggiore, G.; Nastasio, S.; Sciveres, M. Long-term outcome of liver disease-related fibrinogen aguadilla storage disease in a child. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 699. [Google Scholar] [CrossRef]
- Sternin, J.; Choo, R. The power of positive deviancy. An effort to reduce malnutrition in Vietnam offers an important lesson about managing change. Harv. Bus. Rev. 2000, 78, 14–15. [Google Scholar] [PubMed]
- Marsh, D.R.; Schroeder, D.G.; Dearden, K.A.; Sternin, J.; Sternin, M. The power of positive deviance. BMJ 2004, 329, 1177–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callea, F.; Tomà, P.; Bellacchio, E. The Recruitment-Secretory Block (“R-SB”) Phenomenon and Endoplasmic Reticulum Storage Diseases. Int. J. Mol. Sci. 2021, 22, 6807. [Google Scholar] [CrossRef]
- Zhang, X.; Pham, K.; Li, D.; Schutte, R.J.; Hernandez Gonzalo, D.; Zhang, P.; Oshins, R.; Tan, W.; Brantly, M.; Liu, C.; et al. A Novel Small Molecule Inhibits Intrahepatocellular Accumulation of Z-Variant Alpha 1-Antitrypsin In Vitro and In Vivo. Cells 2019, 8, 1586. [Google Scholar] [CrossRef] [Green Version]
| Protein/Disease | α-Syn/PD | AAT/AATD | FG/HHHS |
|---|---|---|---|
| Native Structure | 15 kDa Monomer N-terminal alpha-helix region, a central domain or NAC region and A C-terminal acidic tail | 52 kDa Monomer Nine alpha-helices, two β-sheets and a reactive center loop | 340 kDa triple fibrinogen Aα, Bβ, and γ chains Two lateral globular parts containing the C-terminus of Bβ and γ chains, a central nodule, containing the N-terminus of all chains |
| Polymerization steps | Monomer → dimer → oligomer → fibrils | Monomer → Dimer → Oligomer → Inclusion | Monomer → Oligomer → Protofibril → Fibril |
| Amyloid structure | Amyloid β-sheets in oligomers and fibrils | Amyloid β-sheets in dimers and oligomers | Amyloid fibril protein fibrinogen Aα |
| Inclusion bodies | LBs with more than 90 protein components | Inclusions with dense material and a clear halo in the periphery | Type I: Polygonal shape Type II: Ground glass appearance Type III: Eosinophilic globules, granular structures in the periphery |
| Inclusion proteins | α-syn, Tau protein, ubiquitin, neurofilament protein, β amyloid, among others | AAT M-Z and ZZ alleles | Mutated fibrinogen γ-chain |
| Organelles affected in the cell | α-syn aggregates can be found in all organelles | Only present in the ER | Only present in the ER |
| ER Stress response | UPR Chaperone activation PERK-dependent pathway | IL-6 and IL-8 protein production. Possible UPR activation. ER overload pathway | No available data |
| Organs affected | Across the brain tissue | Liver and lungs | Liver and lungs |
| Onset of disease | Chronic: Duplication/Triplication of SNCA: Symptoms from the age of 40 Idiopathic: From the age of 55 | Chronic: Symptoms from early childhood | Chronic: Symptoms from early childhood or adulthood |
| Hereditary Hypofibrinogenemia with Hepatic Storage | ||||
|---|---|---|---|---|
| Target | Strategy | Results * | Conclusions | Ref. |
| Autophagy | Expression of mutant γD domain of fibrinogen in yeast model | ↑ Clearance of FG in ER by autophagy system | Aggregates of FG are cleared from the ER via the autophagic pathway. | [126] |
| Autophagy | Response to carbamazepine (CBZ) in patients with Fibrinogen storage disease (FSD). | ↑ Autophagic activity by number of autophagocytic vacuoles ↓ Levels of alanine aminotransferase ↓ Caspase and cytokeratin fragments (M30 and M65). | CBZ enhanced autophagy and reduce aggregate-related toxicity in FSD | [138] |
| Proteolytic pathway | Treatment with ursodeoxycholic acid and α-tocopherol in children-patients with aguadilla HFSD | ↓ Aspartate aminotransferase ↓ Alanine aminotransferase ↓ Serum bile acids ↓ Liver damage and fibrosis | This treatment has been proposed in children with HFSD and evidence of liver damage | [257] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padilla-Godínez, F.J.; Ramos-Acevedo, R.; Martínez-Becerril, H.A.; Bernal-Conde, L.D.; Garrido-Figueroa, J.F.; Hiriart, M.; Hernández-López, A.; Argüero-Sánchez, R.; Callea, F.; Guerra-Crespo, M. Protein Misfolding and Aggregation: The Relatedness between Parkinson’s Disease and Hepatic Endoplasmic Reticulum Storage Disorders. Int. J. Mol. Sci. 2021, 22, 12467. https://doi.org/10.3390/ijms222212467
Padilla-Godínez FJ, Ramos-Acevedo R, Martínez-Becerril HA, Bernal-Conde LD, Garrido-Figueroa JF, Hiriart M, Hernández-López A, Argüero-Sánchez R, Callea F, Guerra-Crespo M. Protein Misfolding and Aggregation: The Relatedness between Parkinson’s Disease and Hepatic Endoplasmic Reticulum Storage Disorders. International Journal of Molecular Sciences. 2021; 22(22):12467. https://doi.org/10.3390/ijms222212467
Chicago/Turabian StylePadilla-Godínez, Francisco J., Rodrigo Ramos-Acevedo, Hilda Angélica Martínez-Becerril, Luis D. Bernal-Conde, Jerónimo F. Garrido-Figueroa, Marcia Hiriart, Adriana Hernández-López, Rubén Argüero-Sánchez, Francesco Callea, and Magdalena Guerra-Crespo. 2021. "Protein Misfolding and Aggregation: The Relatedness between Parkinson’s Disease and Hepatic Endoplasmic Reticulum Storage Disorders" International Journal of Molecular Sciences 22, no. 22: 12467. https://doi.org/10.3390/ijms222212467
APA StylePadilla-Godínez, F. J., Ramos-Acevedo, R., Martínez-Becerril, H. A., Bernal-Conde, L. D., Garrido-Figueroa, J. F., Hiriart, M., Hernández-López, A., Argüero-Sánchez, R., Callea, F., & Guerra-Crespo, M. (2021). Protein Misfolding and Aggregation: The Relatedness between Parkinson’s Disease and Hepatic Endoplasmic Reticulum Storage Disorders. International Journal of Molecular Sciences, 22(22), 12467. https://doi.org/10.3390/ijms222212467